Sequence Alignment Map (SAM) is a text-based format originally for storing biological sequences aligned to a reference sequence developed by Heng Li and Bob Handsaker et al.
SAM文件
Sequence Alignment Map(SAM)存储reads到参考基因组比对信息的文本格式。
SAM内容分为两部分,分别是注释信息(head section)和比对结果(Alignment section)。

注释信息
注释信息以@开头
| 缩写 | 含义 |
|---|---|
| @HD | VN: 格式版本; SO: 比对排列顺序 |
| @SQ | 参考序列信息 |
| @RG | Read group信息 |
| @PG | Program, 命令详情 |
| @CO | 其它补充说明 |
比对结果
11个必须字段 + 1个可选字段,字段间TAB分割
| 字段 | 含义 |
|---|---|
| QNAME | FASTQ中的READ ID |
| FLAG | 位标识符 |
| RNAME | 参考序列名称 |
| POS | 1-based比对位置 |
| MAPQ | 比对质量 |
| CIGAR | 简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report) |
| RNEXT | 配对片段(mate)比对上的参考序列的编号,没有另外的片段,这里是’*‘,同一个片段,用’=’ |
| PNEXT | 配对片段(mate)比对到参考序列上的第一个碱基位置,若无mate,则为0; |
| TLEN | Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0 |
| SEQ | reads的序列信息 |
| QUAL | reads的质量信息 |
| Optional fields | 可选字段 |
FLAG位标识符
/*! @abstract the read is paired in sequencing, no matter whether it is mapped in a pair */
#define BAM_FPAIRED 1
/*! @abstract the read is mapped in a proper pair */
#define BAM_FPROPER_PAIR 2
/*! @abstract the read itself is unmapped; conflictive with BAM_FPROPER_PAIR */
#define BAM_FUNMAP 4
/*! @abstract the mate is unmapped */
#define BAM_FMUNMAP 8
/*! @abstract the read is mapped to the reverse strand */
#define BAM_FREVERSE 16
/*! @abstract the mate is mapped to the reverse strand */
#define BAM_FMREVERSE 32
/*! @abstract this is read1 */
#define BAM_FREAD1 64
/*! @abstract this is read2 */
#define BAM_FREAD2 128
/*! @abstract not primary alignment */
#define BAM_FSECONDARY 256
/*! @abstract QC failure */
#define BAM_FQCFAIL 512
/*! @abstract optical or PCR duplicate */
#define BAM_FDUP 1024
/*! @abstract supplementary alignment */
#define BAM_FSUPPLEMENTARY 2048
| 位标识符 | 含义 |
|---|---|
| 1 | PE双端测序 |
| 2 | Read mapped in proper pair, Read和MATE都比对上,且间距合理 |
| 4 | Read没有比对上 |
| 8 | MATE没有比对上 |
| 16 | READ比对到反义链 |
| 32 | MATE比对到反义链 |
| 64 | 这条read是read1 |
| 128 | 这条read是read2 |
| 256 | 不是主要比对(primary alignment) |
| 512 | QC失败 |
| 1024 | PCR重复 |
| 2048 | supplementary alignment |
CIGAR简要比对信息表达式

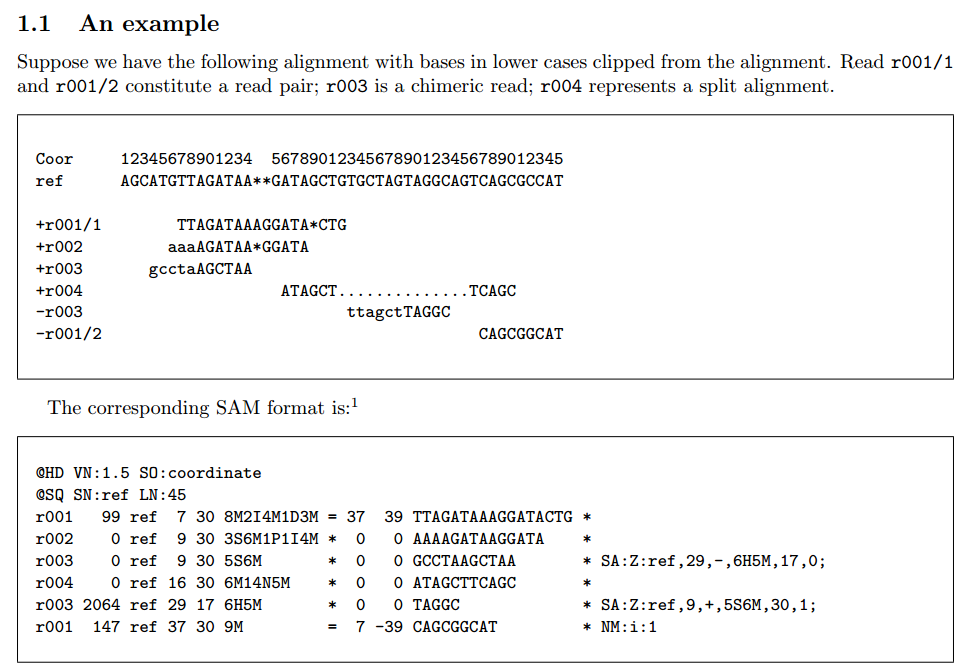
BAM文件
BAM文件是SAM文件的压缩二进制版本,压缩率在5左右。
查看BAM文件
samtools view sample.bam
检查BAM文件完整性
samtools quickcheck sample.bam
## or
tail -c 28 sample.bam | xxd -p
## Output: 1f8b08040000000000ff0600424302001b0003000000000000000000
