7 表达矩阵质控
7.1 UMI表达质控
7.1.1 介绍
基因表达定量后整理为表达矩阵文件,其中每行对应基因(转录本),每列对应单个细胞。检查矩阵去除read QC或mapping QC中低质量细胞,否则会引入技术噪音,模糊下游感兴趣的生物信号。
目前没有通用的scRNA-seq标准化方法,下文中不同质控期望值因不同实验差异很大。因此,质控时,通过比较数据集内部找到异常细胞,而不是独立的质控标准。当比较不同protocol的数据集的质控指标时,应格外注意。
7.1.2 Tung数据集
使用芝加哥大学Yoav Gilad实验室三个不同个体诱导多能干细胞数据集 (Tung et al. 2017)。细胞分选采用Fluidigm C1平台,同时使用UMI和ERCC spike-in,数据文件位于工作目录下的tung文件夹中,这些文件15/03/16创建原始文件的拷贝。
导入数据和注释:
molecules <- read.table("data/tung/molecules.txt", sep = "\t")
anno <- read.table("data/tung/annotation.txt", sep = "\t", header = TRUE)查看表达矩阵
## NA19098.r1.A01 NA19098.r1.A02 NA19098.r1.A03
## ENSG00000237683 0 0 0
## ENSG00000187634 0 0 0
## ENSG00000188976 3 6 1
## ENSG00000187961 0 0 0
## ENSG00000187583 0 0 0
## ENSG00000187642 0 0 0## individual replicate well batch sample_id
## 1 NA19098 r1 A01 NA19098.r1 NA19098.r1.A01
## 2 NA19098 r1 A02 NA19098.r1 NA19098.r1.A02
## 3 NA19098 r1 A03 NA19098.r1 NA19098.r1.A03
## 4 NA19098 r1 A04 NA19098.r1 NA19098.r1.A04
## 5 NA19098 r1 A05 NA19098.r1 NA19098.r1.A05
## 6 NA19098 r1 A06 NA19098.r1 NA19098.r1.A06数据包括 3 个个体,3次3 重复,共 9 个批次.
使用SingleCellExperiment (SCE)和scater标准化分析。首先创建SCE对象:
移除在任何细胞都不表达的基因:
定义control特征(基因) - ERCC spike-ins 和线粒体基因,(作者提供):
isSpike(umi, "ERCC") <- grepl("^ERCC-", rownames(umi))
isSpike(umi, "MT") <- rownames(umi) %in%
c("ENSG00000198899", "ENSG00000198727", "ENSG00000198888",
"ENSG00000198886", "ENSG00000212907", "ENSG00000198786",
"ENSG00000198695", "ENSG00000198712", "ENSG00000198804",
"ENSG00000198763", "ENSG00000228253", "ENSG00000198938",
"ENSG00000198840")计算质量指标:
7.1.3 细胞质控
7.1.3.1 文库大小
查看每个样本检测的总RNA分子数,read counts 或 UMI counts。如果样本中reads/分子数太少,可能细胞破损或捕获失败,应该移除该样本。
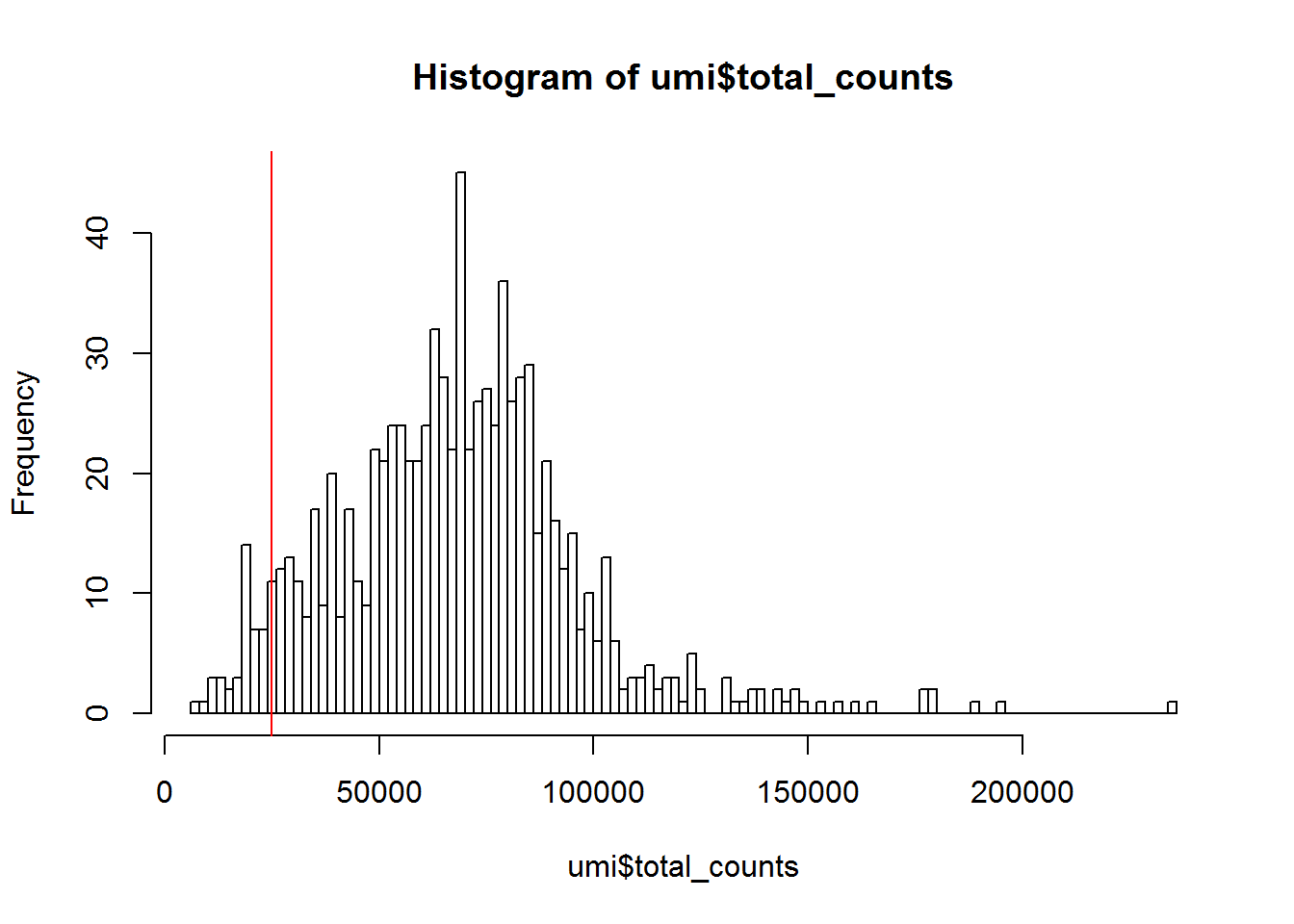
Figure 7.1: Histogram of library sizes for all cells
练习1
过滤移除多少细胞?
每个细胞总分子数应该服从什么分布?
答案
## filter_by_total_counts
## FALSE TRUE
## 46 8187.1.3.2 检测基因数
除了确保每个样品的足够测序深度外,还希望确保读数均衡分布在转录组中。 因此,计算每个样品中检测到的基因数。
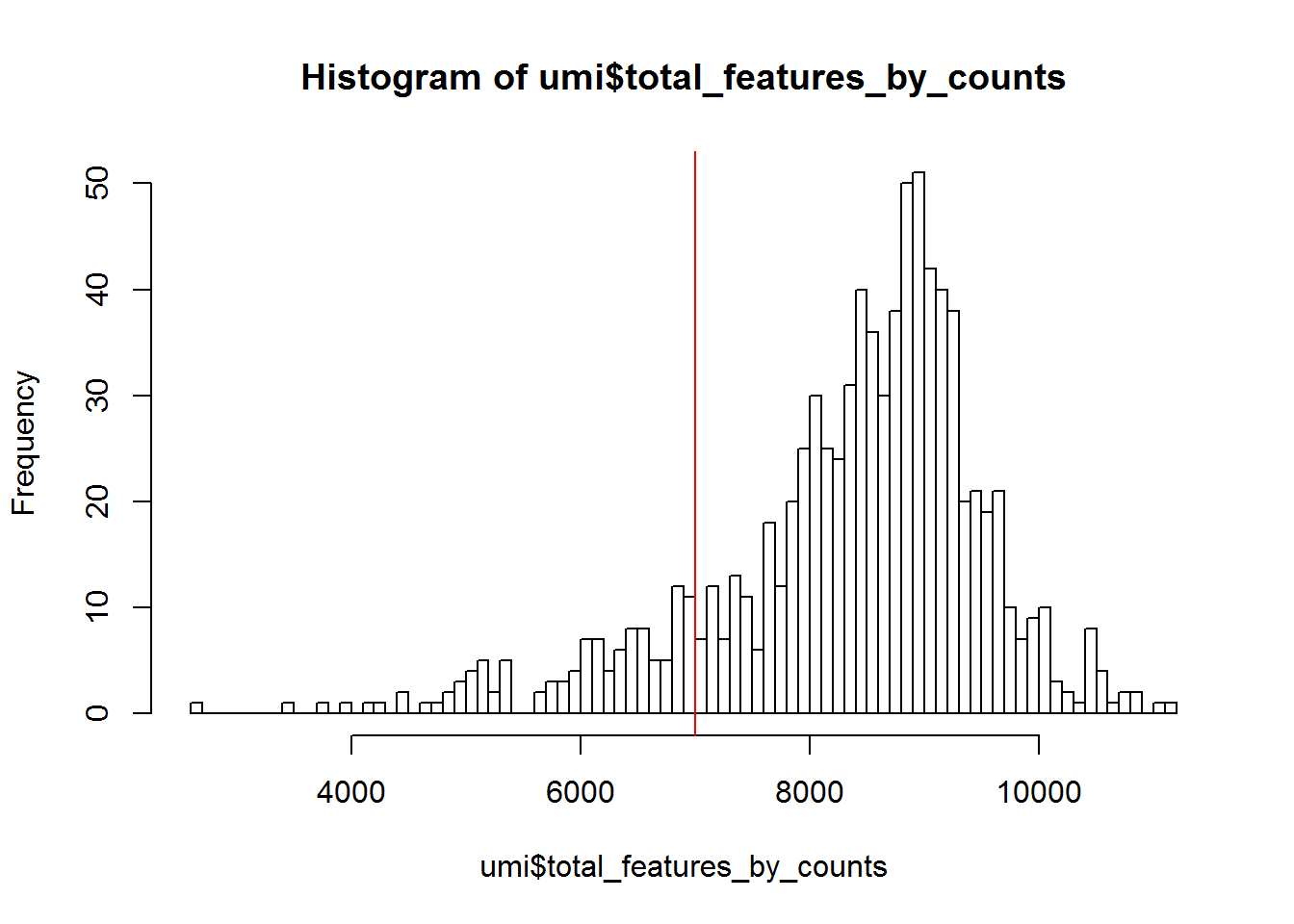
Figure 7.2: Histogram of the number of detected genes in all cells
从上图可以看出大多数细胞检测到7,000-10,000个基因,这对高深度scRAN-seq是正常的。然而,受实验protocol和测序深度的影响。比如基于droplet的方法或样品测序深度低时每个细胞检测基因要少。上图最明显的特征是左侧拖尾严重,如果细胞间检测率相同,应该服从正态分布。因此移除分布在左侧尾部的数据(检测少于7000基因的细胞)
联系2
上述过滤了多少细胞?
答案
## filter_by_expr_features
## FALSE TRUE
## 116 7487.1.3.3 ERCCs和MTs
细胞质量的另一个衡量标准是ERCC spike-in RNA和内源RNA之间的比值。其可用于估计细胞中捕获RNA的总量。 如果spike-in RNA较高,表明细胞内源RNA总量低,可能是由于细胞死亡或受到应激导致RNA降解。 Cells with a high level of spike-in RNAs had low starting amounts of RNA, likely due to the cell being dead or stressed which may result in the RNA being degraded.
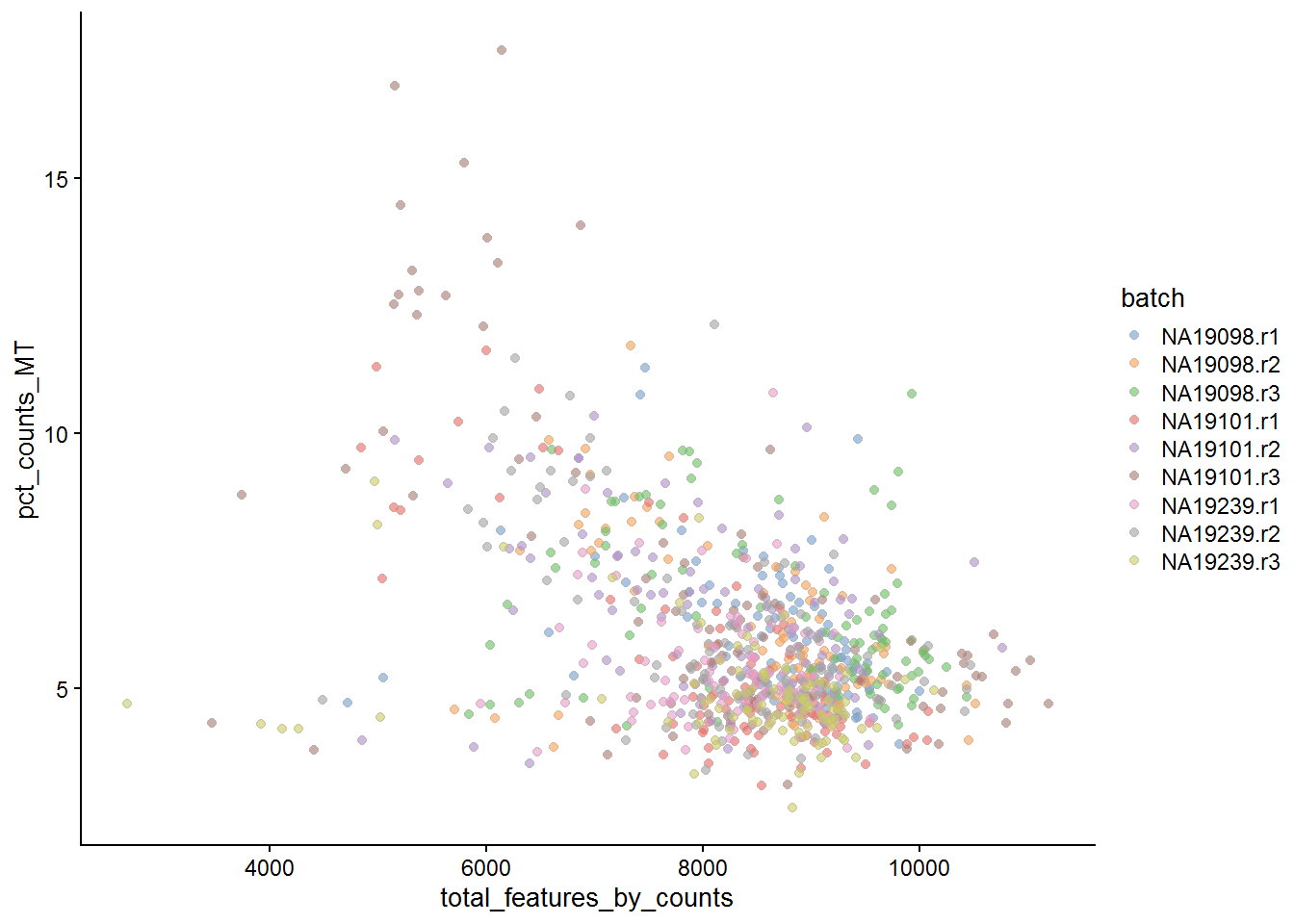
Figure 7.3: Percentage of counts in MT genes
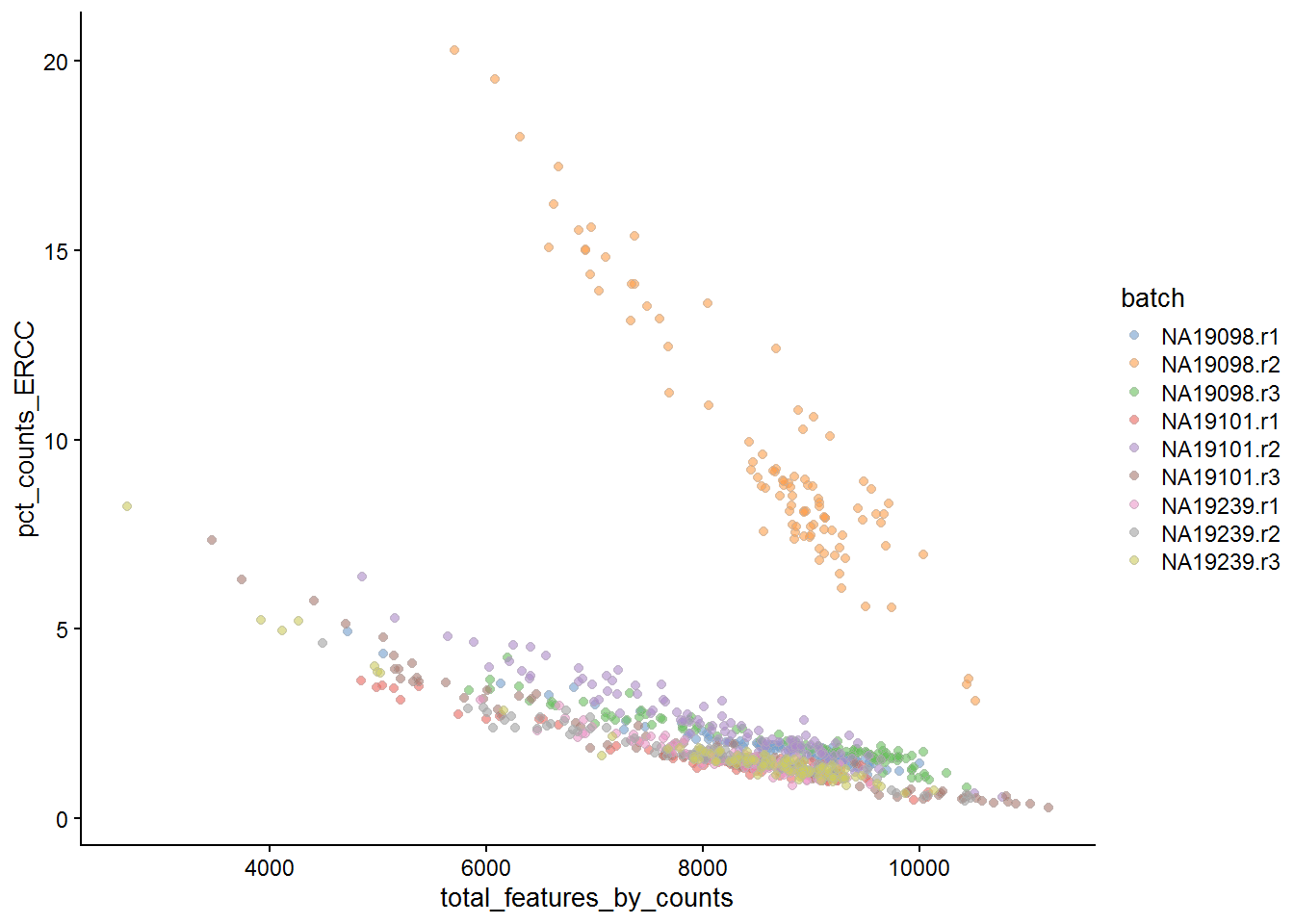
Figure 7.4: Percentage of counts in ERCCs
上述分析表明,来自NA19098.r2批次的大多数细胞具有非常高的ERCC / Endo 比。作者已经解释该批次包含较小尺寸的细胞。
练习3
移除NA19098.r2批次以及移除高表达线粒体基因的细胞(> 10%细胞总计数)
Our answer
## filter_by_ERCC
## FALSE TRUE
## 96 768## filter_by_MT
## FALSE TRUE
## 31 833练习4
如果研究数据集细胞大小不同(比如正常和衰老细胞),ERCC和counts比例会是什么分布?
答案
小的细胞(正常细胞)比大的细胞(衰老细胞)具有更高的ERCC/counts比。
7.1.4 细胞过滤
7.1.4.1 手动过滤
根据之前分析定义细胞过滤器:
umi$use <- (
# sufficient features (genes)
filter_by_expr_features &
# sufficient molecules counted
filter_by_total_counts &
# sufficient endogenous RNA
filter_by_ERCC &
# remove cells with unusual number of reads in MT genes
filter_by_MT
)##
## FALSE TRUE
## 207 6577.1.4.2 自动过滤
scater提供根据质控数据进行PCA自动筛选异常细胞的方法。默认情况下,下列统计量用于基于PCA的异常细胞检测:
- pct_counts_top_100_features
- total_features
- pct_counts_feature_controls
- n_detected_feature_controls
- log10_counts_endogenous_features
- log10_counts_feature_controls
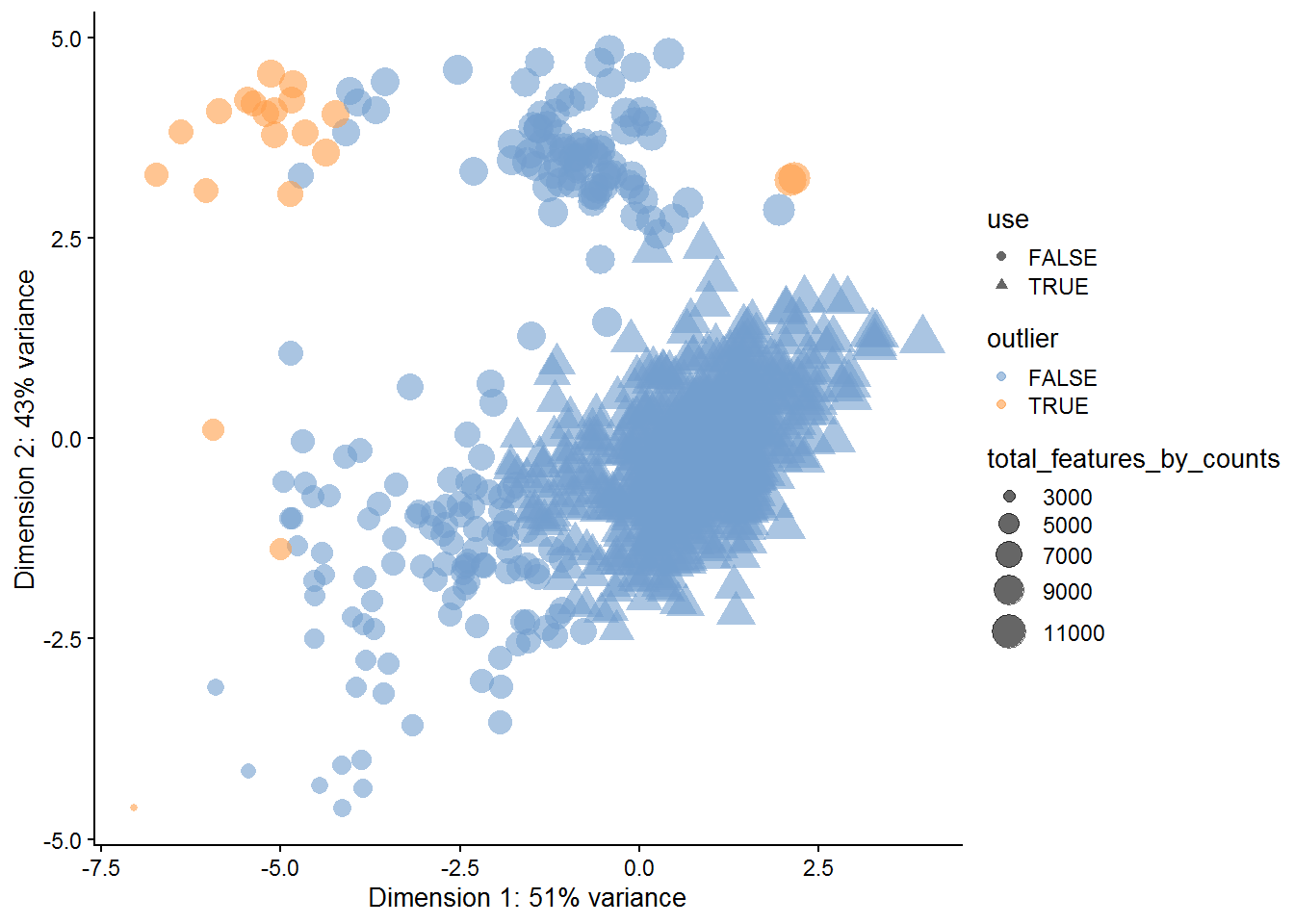
scater首先创建一个行为细胞,列为不同QC统计值的矩阵,然后通过mvoutlier包筛选QC统计值与其它细胞显著不同的异常细胞,可能对应低质量细胞。通过PCA画图可视化异常细胞:
## [1] "PCA_coldata"结果存储于umi的$outlier,其标识细胞是否为异常细胞。自动异常细胞检测提供丰富的信息,但是推荐特异性手动检测过滤数据集。
##
## FALSE TRUE
## 843 21通过PCA查看细胞质量分布
plotReducedDim(
umi,
use_dimred = "PCA_coldata",
size_by = "total_features_by_counts",
shape_by = "use",
colour_by = "outlier"
)
7.1.5 手工过滤和自动过滤比较
练习5
用Venn图显示自动和手工筛选的异常细胞
提示:使用limma中vennCounts和vennDiagram函数绘制Venn图。
答案
##
## 载入程辑包:'limma'## The following object is masked from 'package:scater':
##
## plotMDS## The following object is masked from 'package:BiocGenerics':
##
## plotMAauto <- colnames(umi)[umi$outlier]
man <- colnames(umi)[!umi$use]
venn.diag <- vennCounts(
cbind(colnames(umi) %in% auto,
colnames(umi) %in% man)
)
vennDiagram(
venn.diag,
names = c("Automatic", "Manual"),
circle.col = c("blue", "green")
)
Figure 7.5: Comparison of the default, automatic and manual cell filters
7.1.6 基因分析
7.1.6.1 基因表达
除了移除低质量细胞外,通常也移除受技术误差影响较大的基因。而且查看基因表达谱可以帮助改进实验步骤。
查看Top50表达基因占reads的比例

Figure 7.6: Number of total counts consumed by the top 50 expressed genes
Top50表达的基因reads分布相对平缓,表明(但不保证)细胞的转录组覆盖较好。然而在Top15基因中含有spike-ins,表明如果重复实验,稀释spike-in的浓度较好。
7.1.6.2 基因过滤
通常建议移除表达水平低,被认为是未检测出的基因。UMI数据中基因detectable定义为至少在两个细胞中包含至少1个基因的转录本。read counts数据,基因detectable定义为至少在2个细胞检测到至少5个read比对到该基因上。然而,在两种情况下阈值很大程度上取决于测序深度。另外,基因过滤需在细胞过滤之后,因为一些基因可能只在低质量细胞中检测到。(注意 colData(umi)$use应用于umi数据集)。
keep_feature <- nexprs(
umi[,colData(umi)$use],
byrow = TRUE,
detection_limit = 1
) >= 2
rowData(umi)$use <- keep_feature## keep_feature
## FALSE TRUE
## 4660 14066根据细胞类型,实验protocol,测序深度,其它阈值可能也合适。
7.1.7 保存数据
质控后数据集的维度(注意上述运用的基因过滤):
## [1] 14066 657创建log变换的counts值(下面章节用到),从reducedDim移除保存的PCA结果:
保存数据:
7.1.8 大作业
使用相同的Blischak数据完成质控分析,使用tung/reads.txt文件读入reads,完成后将结果和我们进行对比(下一章)。
7.2 Reads表达质控
reads <- read.table("data/tung/reads.txt", sep = "\t")
anno <- read.table("data/tung/annotation.txt", sep = "\t", header = TRUE)## NA19098.r1.A01 NA19098.r1.A02 NA19098.r1.A03
## ENSG00000237683 0 0 0
## ENSG00000187634 0 0 0
## ENSG00000188976 57 140 1
## ENSG00000187961 0 0 0
## ENSG00000187583 0 0 0
## ENSG00000187642 0 0 0## individual replicate well batch sample_id
## 1 NA19098 r1 A01 NA19098.r1 NA19098.r1.A01
## 2 NA19098 r1 A02 NA19098.r1 NA19098.r1.A02
## 3 NA19098 r1 A03 NA19098.r1 NA19098.r1.A03
## 4 NA19098 r1 A04 NA19098.r1 NA19098.r1.A04
## 5 NA19098 r1 A05 NA19098.r1 NA19098.r1.A05
## 6 NA19098 r1 A06 NA19098.r1 NA19098.r1.A06isSpike(reads, "ERCC") <- grepl("^ERCC-", rownames(reads))
isSpike(reads, "MT") <- rownames(reads) %in%
c("ENSG00000198899", "ENSG00000198727", "ENSG00000198888",
"ENSG00000198886", "ENSG00000212907", "ENSG00000198786",
"ENSG00000198695", "ENSG00000198712", "ENSG00000198804",
"ENSG00000198763", "ENSG00000228253", "ENSG00000198938",
"ENSG00000198840")reads <- calculateQCMetrics(
reads,
feature_controls = list(
ERCC = isSpike(reads, "ERCC"),
MT = isSpike(reads, "MT")
)
)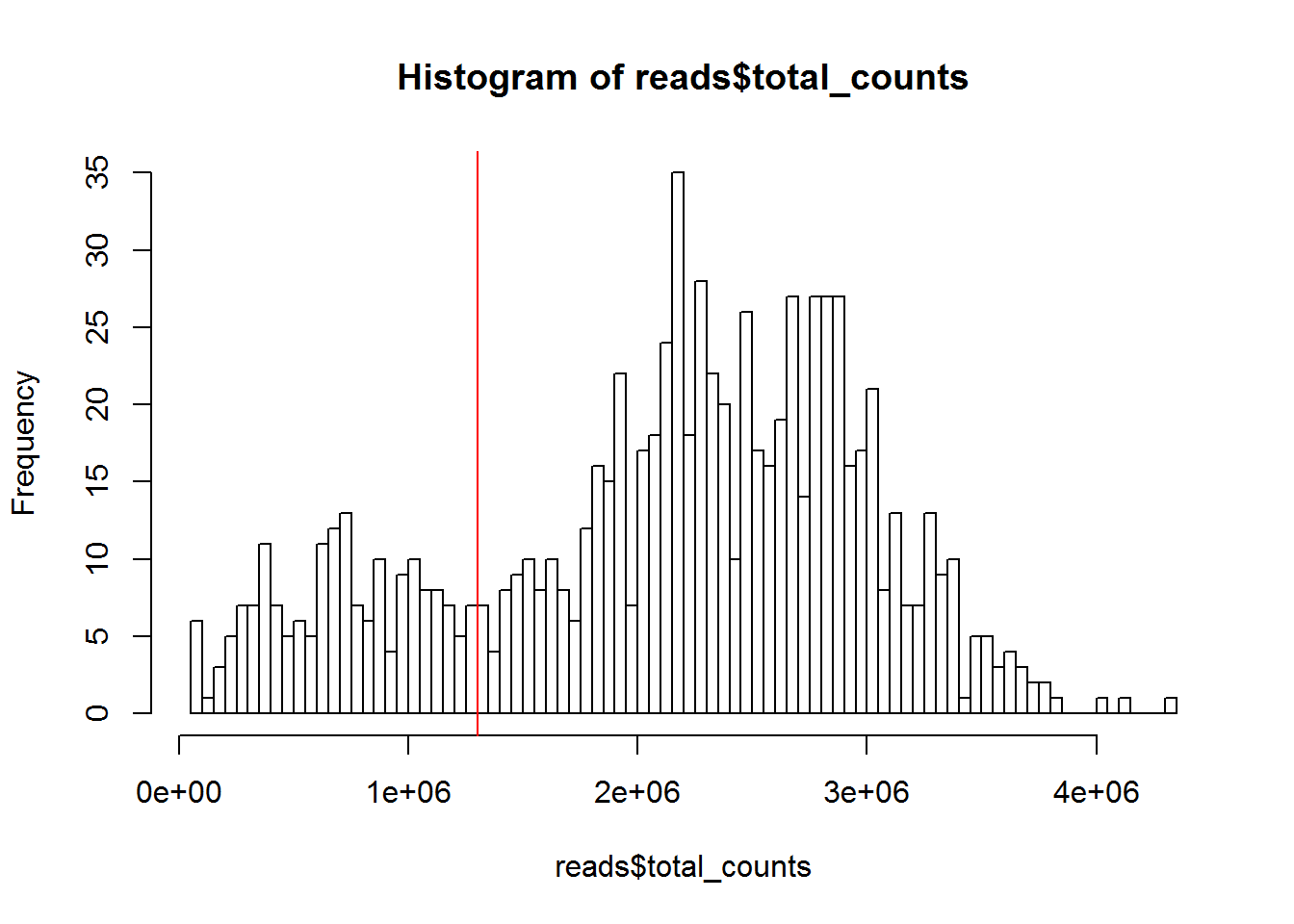
Figure 7.7: Histogram of library sizes for all cells
## filter_by_total_counts
## FALSE TRUE
## 180 684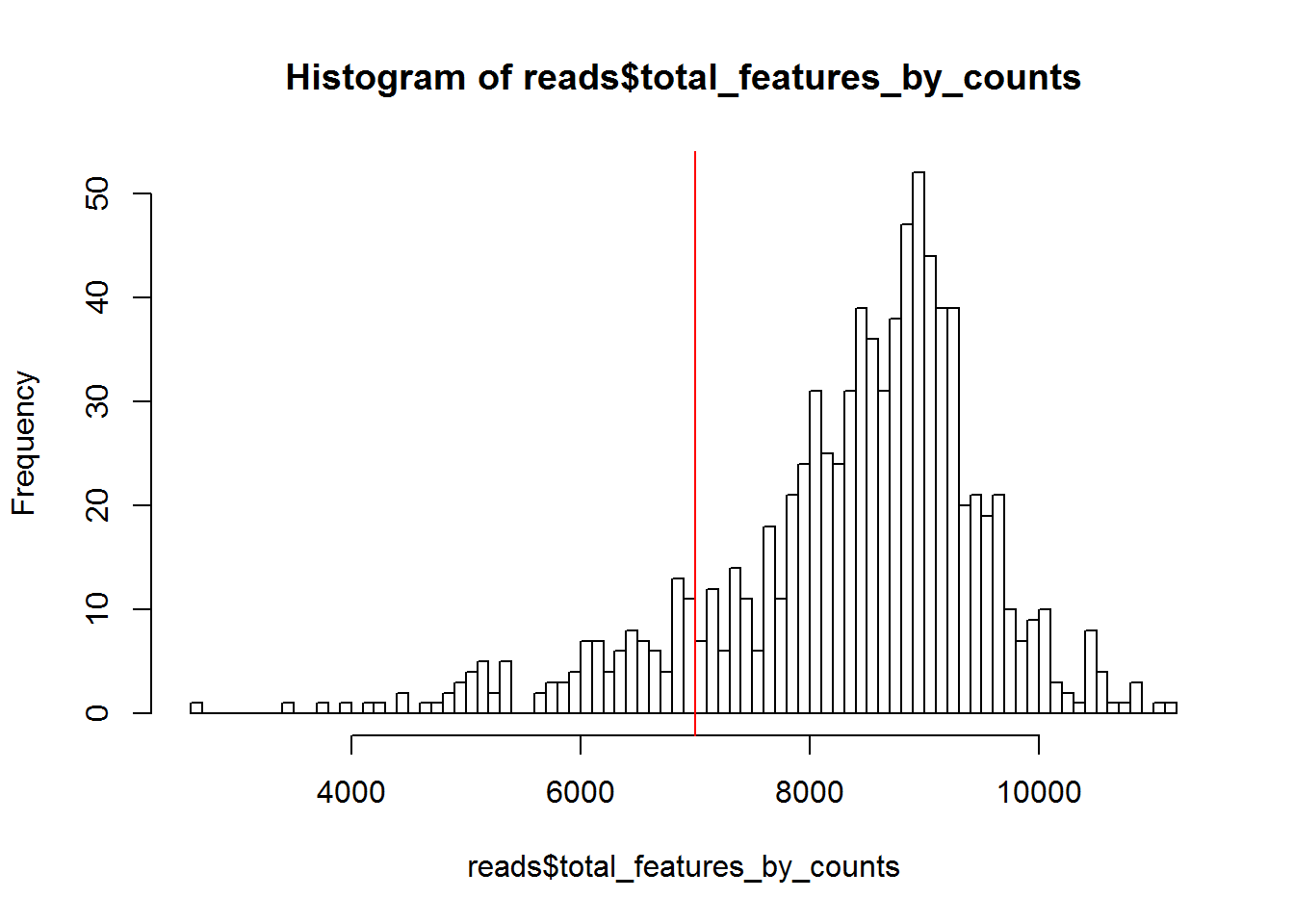
Figure 7.8: Histogram of the number of detected genes in all cells
## filter_by_expr_features
## FALSE TRUE
## 116 748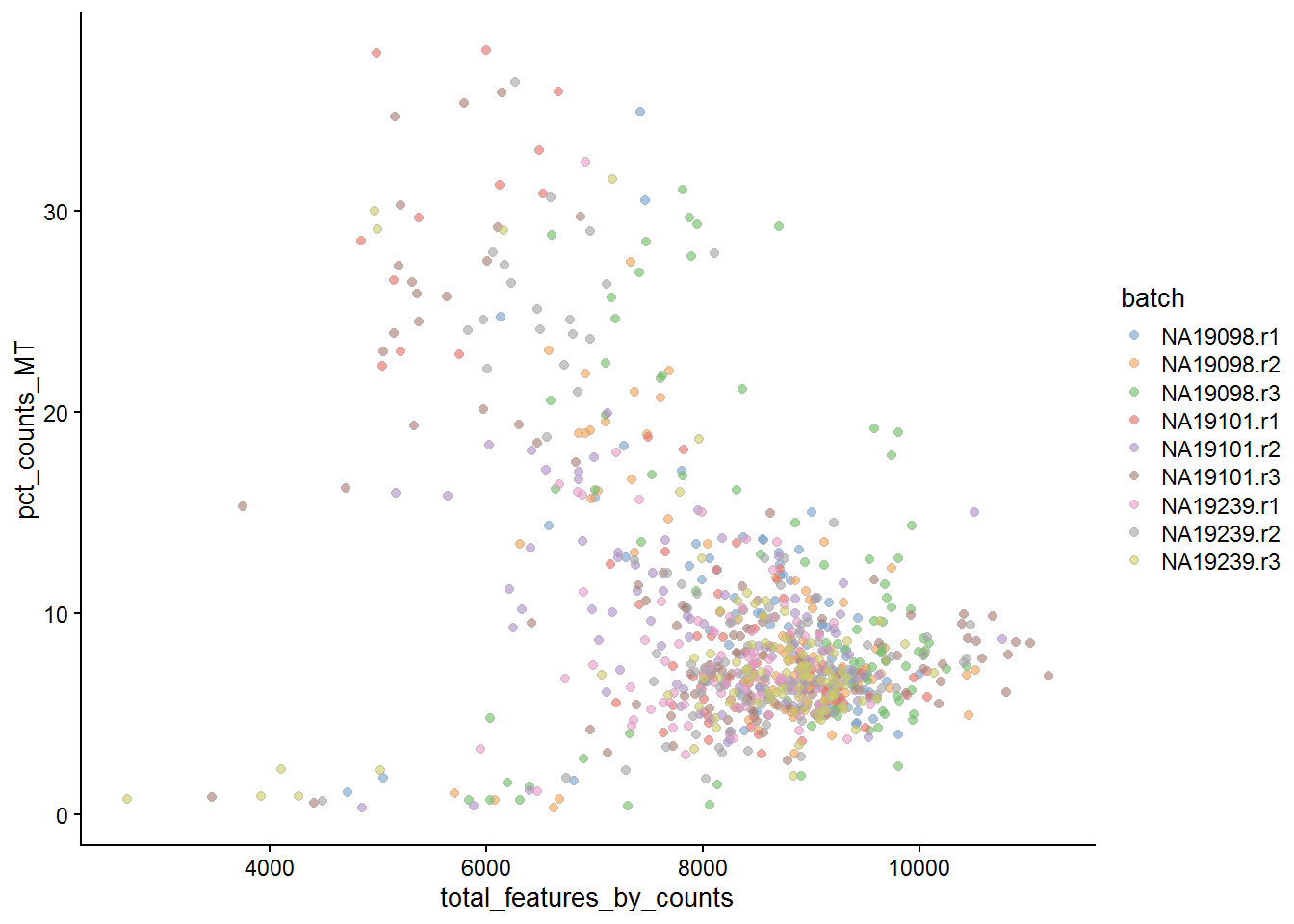
Figure 7.9: Percentage of counts in MT genes
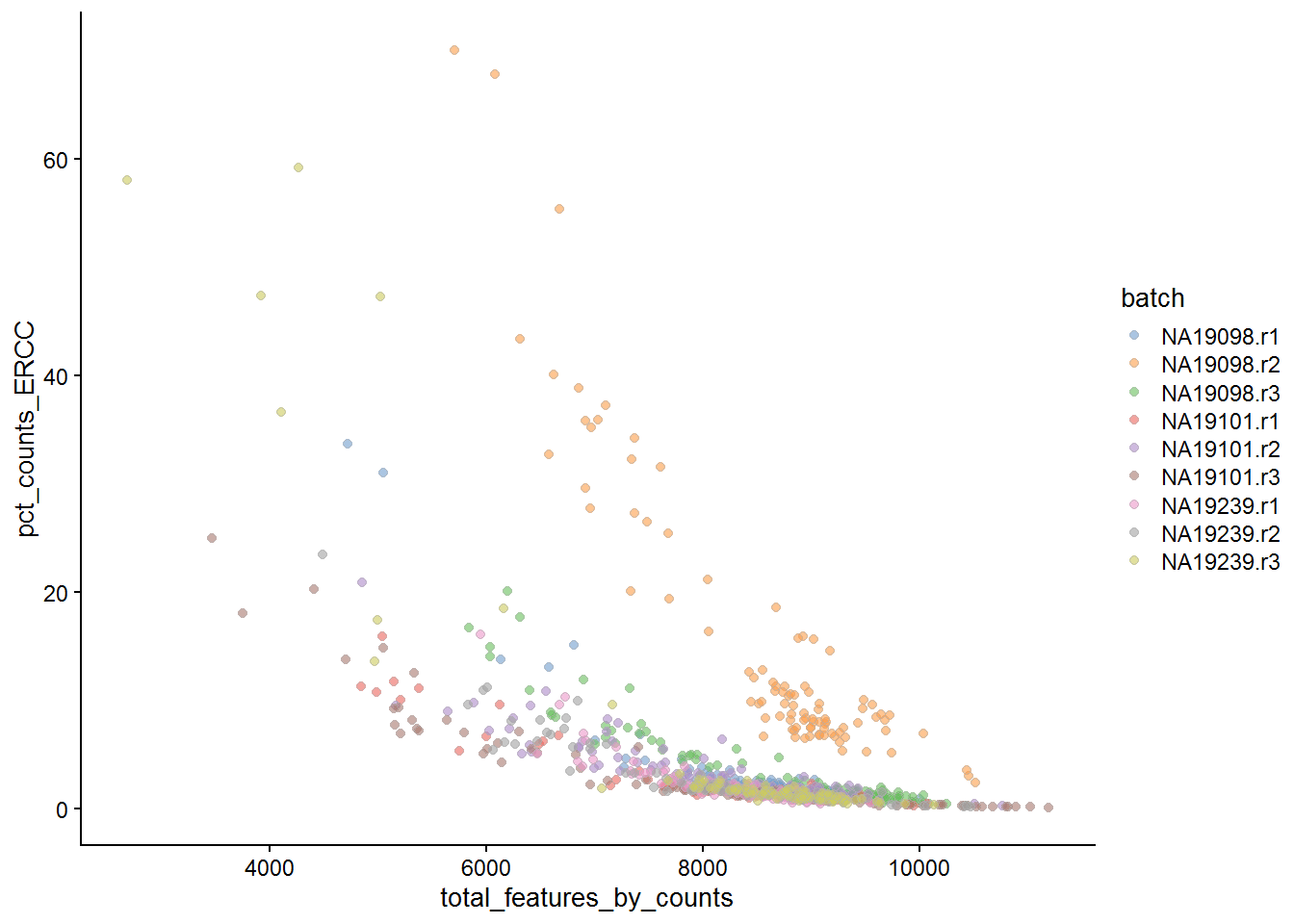
Figure 7.10: Percentage of counts in ERCCs
## filter_by_ERCC
## FALSE TRUE
## 103 761## filter_by_MT
## FALSE TRUE
## 18 846reads$use <- (
# sufficient features (genes)
filter_by_expr_features &
# sufficient molecules counted
filter_by_total_counts &
# sufficient endogenous RNA
filter_by_ERCC &
# remove cells with unusual number of reads in MT genes
filter_by_MT
)##
## FALSE TRUE
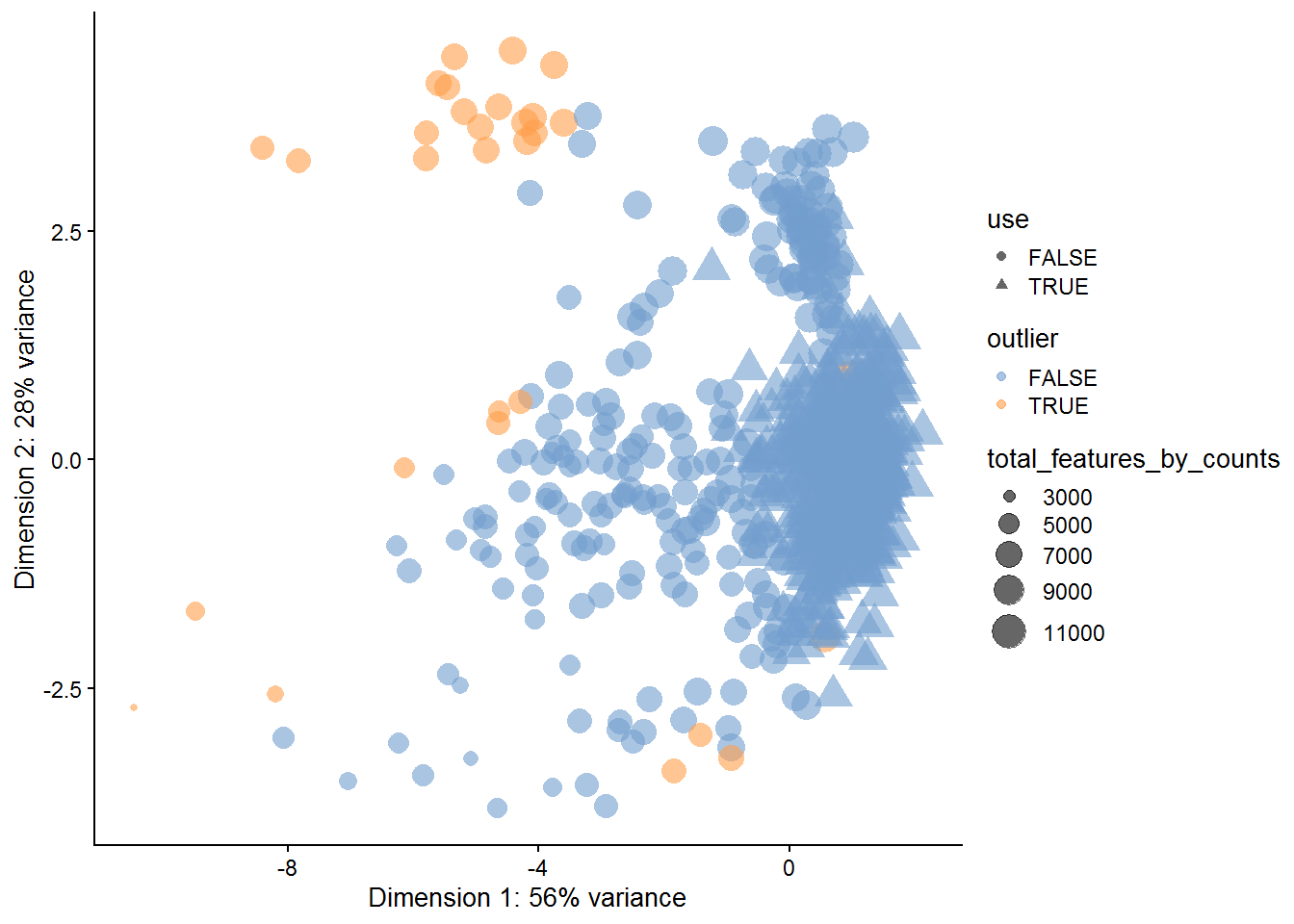
## 258 606## [1] "PCA_coldata"##
## FALSE TRUE
## 834 30plotReducedDim(
reads,
use_dimred = "PCA_coldata",
size_by = "total_features_by_counts",
shape_by = "use",
colour_by = "outlier"
)
library(limma)
auto <- colnames(reads)[reads$outlier]
man <- colnames(reads)[!reads$use]
venn.diag <- vennCounts(
cbind(colnames(reads) %in% auto,
colnames(reads) %in% man)
)
vennDiagram(
venn.diag,
names = c("Automatic", "Manual"),
circle.col = c("blue", "green")
)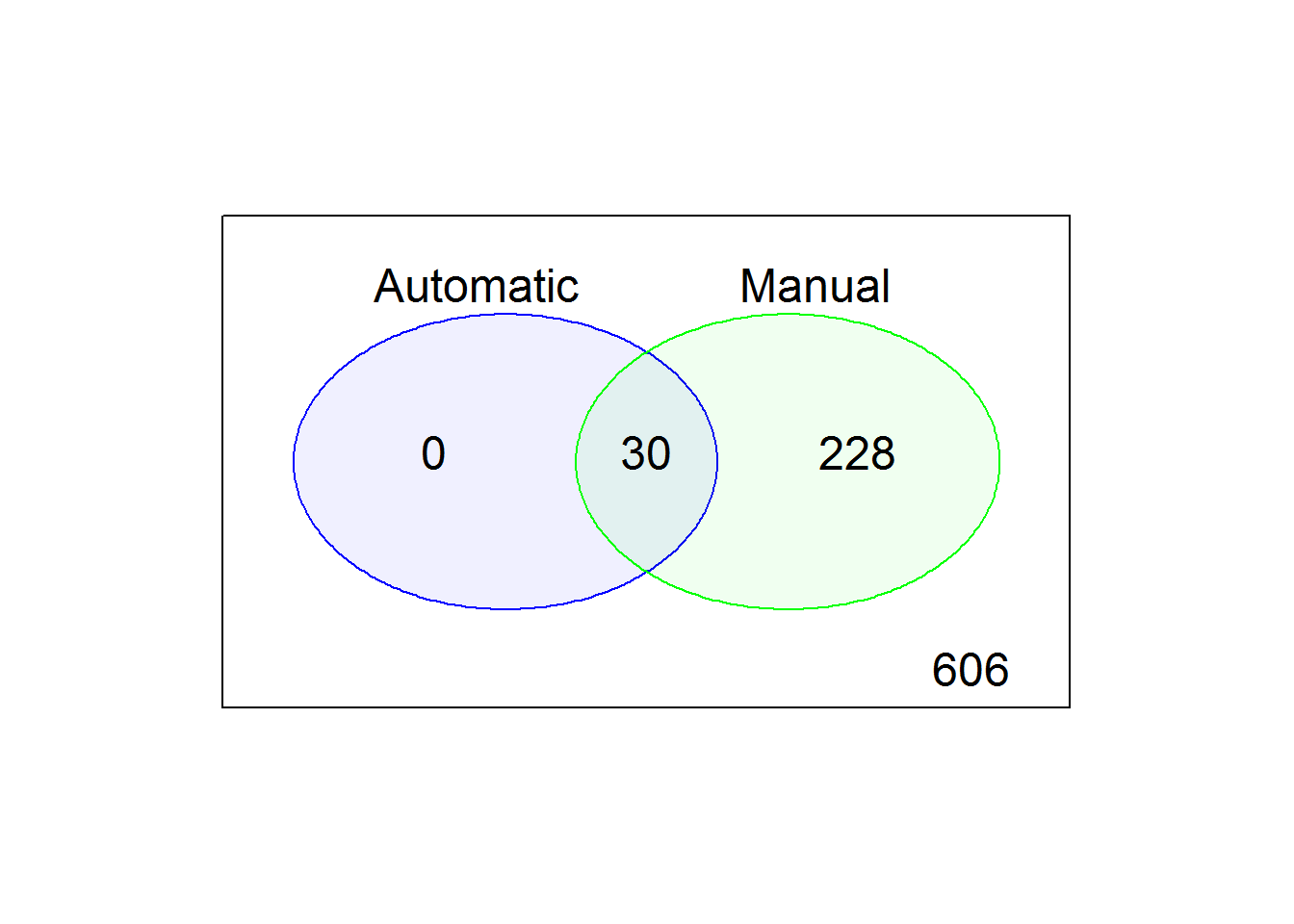
Figure 7.11: Comparison of the default, automatic and manual cell filters

Figure 7.12: Number of total counts consumed by the top 50 expressed genes
keep_feature <- nexprs(
reads[,colData(reads)$use],
byrow = TRUE,
detection_limit = 1
) >= 2
rowData(reads)$use <- keep_feature## keep_feature
## FALSE TRUE
## 2664 16062## [1] 16062 606通过比对图7.5 和 图7.11发现,基于reads过滤比对基于UMI的分析去除了更多的细胞。如果返回比较结果,应该得出结论ERCC和MT过滤对基于reads的分析更严格。
7.3 数据可视化
7.3.1 介绍
本章继续使用之前章节产生的过滤后Tung数据集,我们将使用几种数据可视化方式,以便评估质量控制后表达矩阵的改变。scater包提供了几种有用的函数来简化可视化。
scRNA-sq分析的一个重要方面是控制批次效应。批次效应是实验过程中引入的技术偏差。比如,不同实验室准备的样品或同一实验室不同时间准备的样品,同一批处理的数据相似度更高。最差的情况,批次效应可能被误认为 真实的生物差异。Tung数据详细记录样品处理过程,允许探索批次效应问题。理想情况下,同一个体细胞聚集在一起,不同group对应每个个体,这说明存在批次效应。
7.3.2 PCA plot
查看数据分布的最简单方式是主成分分析,然后查看前两个主成分。
主成分分析(PCA) 是一种统计方法,使用正交变换将观察变量转化为一组线性无关的变量,称之为主成分。主成分的个数少于或等于原始变量数。
数学上,主成分对应协方差矩阵的特征向量,特征向量按照特征值进行排序,使得第一主成分解释最大的数据变异,后续主成分在与前面主成分正交的约下具有最高的方差。(图片来自这里)。
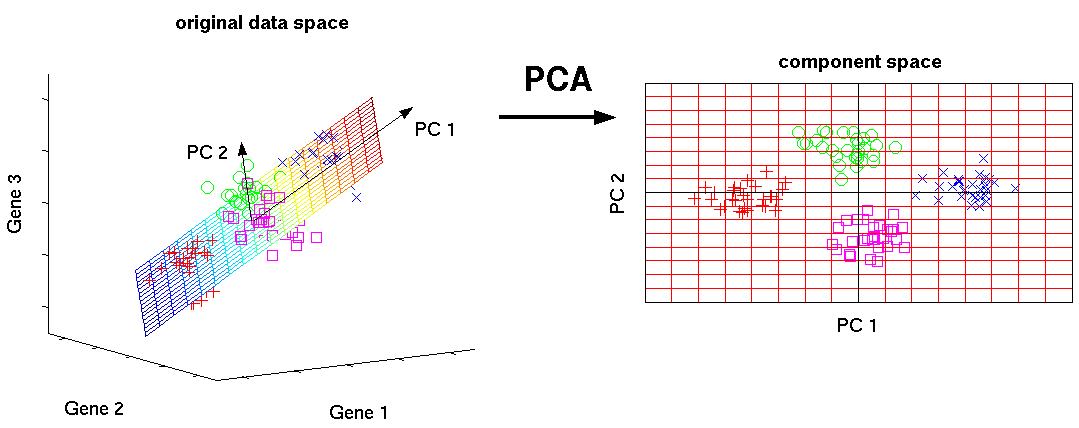
Figure 7.13: Schematic representation of PCA dimensionality reduction
7.3.2.1 Before QC
对数变换前:
tmp <- runPCA(
umi[endog_genes, ],
exprs_values = "counts"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.14: PCA plot of the tung data
对数变换后:
tmp <- runPCA(
umi[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.15: PCA plot of the tung data
显然,对数变换更适合我们的数据,其减少了第一组成分的变异,分离出一些生物效应。而且使表达数据的分布更符合正态分布,后续分析和章节中我们默认使用log变换的counts数据。
然而,仅仅对数变换不足以解释细胞间不同计算因子(如测序深度)带来的差异。因此,下游分析中不要使用logcounts_raw,而是使用SingleCellExperiment对象的logcounts,其不仅仅对数变换,而且根据文库大小进行标准化(比如CPM标准化)。本课程中我们仅使用logcounts_raw进行演示!
7.3.2.2 质控后
tmp <- runPCA(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.16: PCA plot of the tung data
比较图 7.15 和图 7.16, 发现质控后NA19098.r2不再是离群组。
默认情况下,scater使用500个变化最大的基因进行PCA分析,可以通过ntop参数进行修改。
练习1 如果使用所有的14066基因,PCA图会如何?只是用50个基因呢?为什么第一主成分解释整体变异差别那么大?
提示 使用plotPCA函数中ntop参数
答案
tmp <- runPCA(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw",
ntop = 14066
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)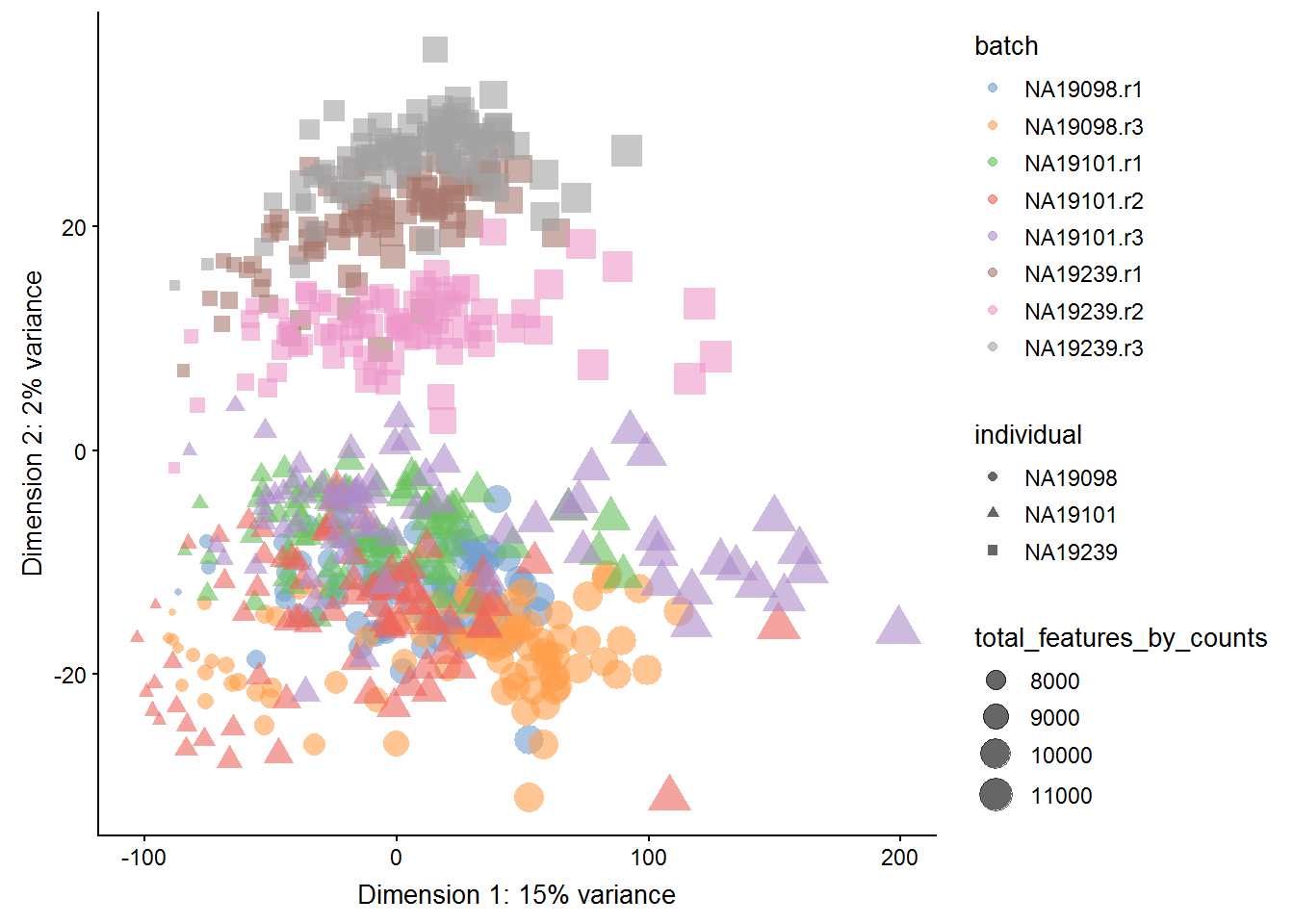
Figure 7.17: PCA plot of the tung data (14214 genes)
tmp <- runPCA(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw",
ntop = 50
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.18: PCA plot of the tung data (50 genes)
7.3.3 tSNE可视化
scRNA-seq数据可视化另一个常用方法是tSNE。tSNE](https://lvdmaaten.github.io/tsne/) (t-Distributed Stochastic Neighbor Embedding)整合降维(比如PCA)和最近邻网络随机游走在保持细胞间局部距离的基础上,将高维数据(比如,我们的14214维表达矩阵)映射到二维空间。和PCA不同的是,tSNE算法具有随机性,即在同一数据集上运行结果可能不同。由于非线性和随机性,tSNE结果难以直观解释。为了确保可重复性,固定随机数“seed”,以便始终得到相同结果。
7.3.3.1 质控前
set.seed(123456)
tmp <- runTSNE(
umi[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 130
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.19: tSNE map of the tung data
7.3.3.2 质控后
set.seed(123456)
tmp <- runTSNE(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 130
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.20: tSNE map of the tung data
因为随机性和非线性,解释PCA和tSNE结果通常较难,不太直观,然而,在这种情况他们提供了相似的数据概览。比较图7.19和图7.20发现,质控过滤后的NA19098.r2样本不再是异常值。
tSNE中perplexity参数表示构建最近邻网络的邻居数,perplexity越高,网络越密集,细胞聚集在一起。perplexity越低,网络越稀疏,细胞群体彼此分离。scater使用默认perplexity为细胞总数除以5(向下取整)。
tSNE的缺点见这里。
练习2 当perplexity设置为10或200时对tSNE结果的影响?
答案
set.seed(123456)
tmp <- runTSNE(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 10
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.21: tSNE map of the tung data (perplexity = 10)
set.seed(123456)
tmp <- runTSNE(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 200
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.22: tSNE map of the tung data (perplexity = 200)
7.3.4 大作业
使用Blischak数据的read counts数据完成相同的分析,使用tung/reads.rds文件导入reads的SCE对象。完成后的结果与我们相比较(下一章)。
7.4 Reads数据可视化
library(scater)
options(stringsAsFactors = FALSE)
reads <- readRDS("data/tung/reads.rds")
reads.qc <- reads[rowData(reads)$use, colData(reads)$use]
endog_genes <- !rowData(reads.qc)$is_feature_controltmp <- runPCA(
reads[endog_genes, ],
exprs_values = "counts"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.23: PCA plot of the tung data
tmp <- runPCA(
reads[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.24: PCA plot of the tung data
tmp <- runPCA(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.25: PCA plot of the tung data
set.seed(123456)
tmp <- runTSNE(
reads[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 130
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.26: tSNE map of the tung data
set.seed(123456)
tmp <- runTSNE(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 130
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.27: tSNE map of the tung data
set.seed(123456)
tmp <- runTSNE(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 10
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.28: tSNE map of the tung data (perplexity = 10)
set.seed(123456)
tmp <- runTSNE(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw",
perplexity = 200
)
plotTSNE(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.29: tSNE map of the tung data (perplexity = 200)
7.5 识别混淆因子
7.5.1 介绍
scRNA-seq数据中存在大量潜在的混淆因子,假象和偏差。分析scRNA-seq数据的一个主要挑战是难以进行真正的技术重复来区分生物变异和技术变异。前面的章节中考虑了批次效应,本章节将继续探索如何识别和移除实验假象。scater包提供了一套专门用于实验和解释变量质控的方法。而且,我们将继续使用上一章节的Blichak数据。
library(scater, quietly = TRUE)
options(stringsAsFactors = FALSE)
umi <- readRDS("data/tung/umi.rds")
umi.qc <- umi[rowData(umi)$use, colData(umi)$use]
endog_genes <- !rowData(umi.qc)$is_feature_controlumi.qc数据集包含过滤后的细胞和基因。下一步将探索数据变异的技术驱动因素,以便在下游分析前进行数据标准化。
7.5.2 和组成分的相关性
首先查看质控后数据集的PCA图:
tmp <- runPCA(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts"
)
Figure 7.30: PCA plot of the tung data
scater可以识别与感兴趣的实验/QC变量相关的主成分,通过构建组成分值和感兴趣变量的回归模型,按照\(R^2\)对主成分进行排序。
测试一些变量是否与主成分相关。
7.5.2.1 检测的基因
logcounts(umi.qc) <- assay(umi.qc, "logcounts_raw")
plotExplanatoryPCs(
umi.qc[endog_genes, ],
variables = "total_features_by_counts"
)
logcounts(umi.qc) <- NULL
Figure 7.31: PC correlation with the number of detected genes
实际上,可以看出PC1几乎可以完全用检测到的基因数来解释。实际上,在上述的PCA图中可以看到,这是scRNA-seq分析中著名的问题,详见这里。
7.5.3 解释变量
scater通过拟合每个基因表达值和相应变量的回归模型计算变量的边际\(R^2\),下图显示变量的基因边际\(R^2\)值的密度图。
plotExplanatoryVariables(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw",
variables = c(
"total_features_by_counts",
"total_counts",
"batch",
"individual",
"pct_counts_ERCC",
"pct_counts_MT"
)
)
Figure 7.32: Explanatory variables
该分析表明,检测到的基因数目以及测序深度(number of counts)对许多基因具有实质性的解释力,因此在标准化步骤或在下游统计模型中需要考虑这些变量。ERCC的表达似乎也是一个重要的解释变量,上图的一个显著特征是批次效应比个体具有更高的解释效力。这告诉我们数据的技术和生物变异是什么?
7.5.4 其它混淆因子
除了校正批次效应,还需要考虑其它因素,这同样需要外部信息。一种流行的方法是scLVM,其识别和去除细胞周期或凋亡等过程的影响。
此外,protocols在转录本覆盖率,A/T平均含量偏差,捕获短转录本效率方面具有差异,理想情况下,希望校正所有误差。
7.5.5 练习
使用Blischak数据中read counts执行相同的分析,使用tung/reads.rds文件导入SCESet对象,完成分析后将结果与我们的相比较(见下一章)。
7.6 识别READS中混淆因子
library(scater, quietly = TRUE)
library(knitr)
options(stringsAsFactors = FALSE)
opts_chunk$set(out.width='90%', fig.align = 'center', echo=FALSE)
reads <- readRDS("data/tung/reads.rds")
reads.qc <- reads[rowData(reads)$use, colData(reads)$use]
endog_genes <- !rowData(reads.qc)$is_feature_controltmp <- runPCA(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts"
)
Figure 7.33: PCA plot of the tung data
logcounts(reads.qc) <- assay(reads.qc, "logcounts_raw")
plotExplanatoryPCs(
reads.qc[endog_genes, ],
variables = "total_features_by_counts"
)
logcounts(reads.qc) <- NULL
Figure 7.34: PC correlation with the number of detected genes
plotExplanatoryVariables(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw",
variables = c(
"total_features_by_counts",
"total_counts",
"batch",
"individual",
"pct_counts_ERCC",
"pct_counts_MT"
)
)
Figure 7.35: Explanatory variables
7.7 标准化理论
7.7.1 介绍
在前一章中,我们识别了重要的混淆因子和解释变量。scater可以在后续统计模型中考虑这些变量,或者如果需要,使用normaliseExprs()来调节它们。这可以通过提供设计矩阵给normaliseExprs()来完成。我们这里没有涉及这个主题,但你可以尝试自己做这个练习。
相反,我们将探索校正文库大小的简单尺寸因子标准化如何消除某些混淆因素和解释变量的影响。
7.7.2 文库大小
因为scRNA-seq数据通常在highly multiplexed平台测序,来自每个细胞的总reads数可能显著不同,从而导致文库大小差异。一些定量方法(比如,Cufflinks, RSEM) 在估计基因表达时考虑文库大小,因此不需要再对文库大小进行标准化。
然而,如果使用其它的定量方法,则必须将表达矩阵中每列乘以或除以“归一化因子”来校正文库大小,其中归一化因子是相对于其它细胞的文库大小估计。用于bulk RNA-seq文库大小校正的方法也可应用于scRNA-seq(比如, UQ, SF, CPM, RPKM, FPKM, TPM).
7.7.3 标准化
7.7.3.1 CPM
数据标准化最简单的方式是将每列除以总数再乘以1,000,000,转化为counts per million(CPM)。注意,校正细胞内RNA总量时,应该排除spike-ins,只使用内源基因。R中CPM函数示例:
calc_cpm <-
function (expr_mat, spikes = NULL)
{
norm_factor <- colSums(expr_mat[-spikes, ])
return(t(t(expr_mat)/norm_factor)) * 10^6
}CPM 一个缺点是如果样本含有在细胞中高表达和差异表达的基因,细胞中的总分子数可能取决于这些基因是否在细胞中开/关状态并且通过总分子数标准化可能隐藏那些基因的差异表达和/或错误地为剩余基因产生差异表达。
注意 RPKM, FPKM 和 TPM 是 CPM 的变形,其根据相应基因/转录本的长度调节counts。
为解决上述问题,设计了其它几种方法:
7.7.3.2 RLE (SF)
尺寸因子(SF)由DESeq (Anders and Huber 2010)提出并推广,首先计算细胞间每个基因的表达几何平均值。每个细胞的尺寸因子是基因表达值与几何均值比值的中位数。该方法的缺点在于,由于它使用几何平均值,因此在所有细胞中仅非零表达的基因可用于计算,使得不适用大规模低深度scRNA-Seq实验。edgeR和scater将此方法称为RLE,“relative log expression”。 R中的SF函数示例:
7.7.3.3 UQ
上四分位数(upperquartil, UQ)由(Bullard et al. 2010)提出。每列除以该文库的75%分位数的表达值。 通常,计算的分位数通过细胞间的中值来缩放,以保持表达的绝对水平相对一致。 该方法的缺点在于,对于低深度scRNA-Seq实验,大量未检测到的基因可能导致75%分位数为零(或接近它)。 可以通过使用更高的分位数(例如,99%分位数是scater中的默认值)或在计算75%分位数之前去除零值来克服该限制。 R中*UQ函数的示例:
7.7.3.4 TMM
TMM是(Robinson and Oshlack 2010)提出的weighted trimmed mean of M-values,M-values为细胞间基因的log2变化倍数,使用一个细胞作为参考,计算每个细胞相比于参考细胞的M值。考虑log变换对方差的影响,去除顶部和底部的30%,计算剩下值的加权均值。每个非参考细胞乘以计算的因子。该方法的两个潜在问题是trimming后非零基因太少;基于大多数基因都不差异表达的假设。
7.7.3.5 scran
scran包实现了用于单细胞数据的CPM方法(L. Lun, Bach, and Marioni 2016)。该方法通过池化细胞计算标准化因子(类似CPM)解决单个细胞中零值非常多的问题。由于每个细胞都存在于不同的池中,因此细胞特异性因子可以使用线性代数从池特定因子的集合中去卷积的得出。
7.7.3.6 下采样(Downsampling)
校正文库大小的另外一种方法是对表达矩阵进行下采样,使每个细胞具有大致相同数目的分子数。该方法的好处是通过下采样引入零值,从而消除由于检测不同数量的基因引起的偏差。然而,主要缺点是该过程不是确定性的,因此每次运行下采样时,得到的表达矩阵略有不同。 因此,通常必须对多次下采样进行分析,以确保结果稳健。R中downsampling函数的示例:
7.7.4 Effectiveness
比较不同保准化方法的效果,使用PCA图进行可视化并通过scatter中plotRLE()函数计算细胞间 相对log表达 。即,具有较多(较少)reads的细胞比大多数基因中间表达水平高(低),导致整个细胞阳性(阴性) RLE。R中 RLE 函数示例:
calc_cell_RLE <-
function (expr_mat, spikes = NULL)
{
RLE_gene <- function(x) {
if (median(unlist(x)) > 0) {
log((x + 1)/(median(unlist(x)) + 1))/log(2)
}
else {
rep(NA, times = length(x))
}
}
if (!is.null(spikes)) {
RLE_matrix <- t(apply(expr_mat[-spikes, ], 1, RLE_gene))
}
else {
RLE_matrix <- t(apply(expr_mat, 1, RLE_gene))
}
cell_RLE <- apply(RLE_matrix, 2, median, na.rm = T)
return(cell_RLE)
}注意, RLE, TMM, 和 UQ 尺寸因子方法是伪bulk RNA-seq数据开发的,依赖于实验情况,可能不适合scRNA-seq数据,因为其基于的假设可能不适用于scRNA-seq。
注意 scater封装了edgeR的calcNormFactors函数,其包括多种文库大小标准化方法,可以非常方便地应用于我们数据。
注意 edgeR对一些标准化方法进行了一些调整,和原始方法运行的结果有些差异,比如edgeR和scater的“RLE”方法基于 DESeq使用的“尺寸因子”,其结果可能和DESeq/DESeq2包中estimateSizeFactorsForMatrix方法结果不同。而且,一些版本的edgeR不会正确计算标准化因子,除非所有细胞的lib.size都设置为1。
注意 对于CPM标准化,使用scater中calculateCPM()函数,对 RLE, UQ 和 TMM,使用scater中normaliseExprs()函数(现已启用,因此删除了相应章节)。对于 scran,我们使用scran包计算尺寸因子(其还在SingleCellExperiment类中运行)和scater的normalize()对数据进行标准化。
所有标准化函数将结果保存在SCE对象中logcounts slot中,对于 downsampling,使用上述自定义函数。
7.8 UMI标准化练习
继续使用上一章节的tung数据集。
library(scRNA.seq.funcs)
library(scater)
library(scran)
options(stringsAsFactors = FALSE)
set.seed(1234567)
umi <- readRDS("data/tung/umi.rds")
umi.qc <- umi[rowData(umi)$use, colData(umi)$use]
endog_genes <- !rowData(umi.qc)$is_feature_control7.8.1 Raw
tmp <- runPCA(
umi.qc[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.36: PCA plot of the tung data
7.8.2 CPM
logcounts(umi.qc) <- log2(calculateCPM(umi.qc, use_size_factors = FALSE) + 1)
plotPCA(
umi.qc[endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.37: PCA plot of the tung data after CPM normalisation

Figure 7.38: Cell-wise RLE of the tung data

Figure 7.39: Cell-wise RLE of the tung data
7.8.3 scran
qclust <- quickCluster(umi.qc, min.size = 30)
umi.qc <- computeSumFactors(umi.qc, sizes = 15, clusters = qclust)
umi.qc <- normalize(umi.qc)
plotPCA(
umi.qc[endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.40: PCA plot of the tung data after LSF normalisation

Figure 7.41: Cell-wise RLE of the tung data

Figure 7.42: Cell-wise RLE of the tung data
scran有时会计算出尺寸因子为负或0,这将完全扭曲标准化的表达矩阵。 我们可以检查scran计算的尺寸因子:
summary(sizeFactors(umi.qc))
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.4832 0.7812 0.9535 1.0000 1.1469 3.2595这个数据集,所有尺寸因子都是合理的。如果发现scran计算的尺寸因子为负,尝试增加cluster和pool的大小直达为正数。
7.8.4 下采样
logcounts(umi.qc) <- log2(Down_Sample_Matrix(counts(umi.qc)) + 1)
plotPCA(
umi.qc[endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.43: PCA plot of the tung data after downsampling

Figure 7.44: Cell-wise RLE of the tung data

Figure 7.45: Cell-wise RLE of the tung data
7.8.5 基因/转录本长度标准化
一些方法结合了文库大小和片段/基因长度进行标准化,比如:
- RPKM - Reads Per Kilobase Million (单端测序)
- FPKM - Fragments Per Kilobase Million (和RPKM相同,针对双端测序, 确保比对到同一片段的paired reads不计算两次)
- TPM - Transcripts Per Kilobase Million (和RPKM相同, 但标准化顺序相反,先对长度进行标准化然后对测序深度)
这些方法不适用于我们的数据集,因为包含UMI的转录本末端更容易被测序,而且,一般来说应该使用合适的定量软件从比对好的BAM文件而不是从read counts计算,因为基因/转录本的一部分进行测序,而不是全长。如果有疑问,查看基因/转录本长度和表达水平的关系。
这里展示如何使用scater进行标准化。首先需要找到有效的转录本长度,然而我们数据集只包括gene IDs,因此使用基因长度而不是转录本。scater使用biomaRt包,可以使用其它属性注释基因。
which allows one to annotate genes by other attributes:
umi.qc <- getBMFeatureAnnos(
umi.qc,
filters = "ensembl_gene_id",
attributes = c(
"ensembl_gene_id",
"hgnc_symbol",
"chromosome_name",
"start_position",
"end_position"
),
biomart = "ENSEMBL_MART_ENSEMBL",
dataset = "hsapiens_gene_ensembl",
host = "www.ensembl.org"
)
# If you have mouse data, change the arguments based on this example:
# getBMFeatureAnnos(
# object,
# filters = "ensembl_transcript_id",
# attributes = c(
# "ensembl_transcript_id",
# "ensembl_gene_id",
# "mgi_symbol",
# "chromosome_name",
# "transcript_biotype",
# "transcript_start",
# "transcript_end",
# "transcript_count"
# ),
# biomart = "ENSEMBL_MART_ENSEMBL",
# dataset = "mmusculus_gene_ensembl",
# host = "www.ensembl.org"
# )移除没有注释到的基因:
使用end_position和start_position字段计算基因长度。

Figure 7.46: Gene length vs Mean Expression for the raw data
基因长度和平均表达值间没有明显关系,因此FPKM和TPM不适合此数据集,但是下面仍展示使用这些方法。
注意 这里计算总基因长度而不是总外显子长度。很多基因包含内含子,因此其eff_length和我们计算的有差异。我们的计算只是个近似,如果使用总外显子长度,参照这篇文章。
进行标准化:
对结果绘制PCA图:
tmp <- runPCA(
umi.qc.ann,
exprs_values = "tpm",
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.47: PCA plot of the tung data after TPM normalisation
tmp <- runPCA(
umi.qc.ann,
exprs_values = "tpm",
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.48: PCA plot of the tung data after FPKM normalisation
注意 PCA寻找细胞间的差异。不同细胞间每个基因长度相同,因此FPKM和CPM图基本一致(只是旋转了一下)因为先计算CPM,然而对基因长度进行标准化。然而TPM不同,因为在计算CPM前先对基因按照长度进行加权。
7.8.6 练习
使用tung数据中read counts完成相同的分析,使用tung/reads.rds加载SCE对象,完成分析后将结果和我们进行比对(下一章)。
7.9 Reads标准化练习
library(scRNA.seq.funcs)
library(scater)
library(scran)
options(stringsAsFactors = FALSE)
set.seed(1234567)
library(knitr)
opts_chunk$set(out.width='90%', fig.align = 'center', echo=FALSE)
reads <- readRDS("data/tung/reads.rds")
reads.qc <- reads[rowData(reads)$use, colData(reads)$use]
endog_genes <- !rowData(reads.qc)$is_feature_controltmp <- runPCA(
reads.qc[endog_genes, ],
exprs_values = "logcounts_raw"
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.49: PCA plot of the tung data
logcounts(reads.qc) <- log2(calculateCPM(reads.qc, use_size_factors = FALSE) + 1)
plotPCA(
reads.qc[endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.50: PCA plot of the tung data after CPM normalisation

Figure 7.51: Cell-wise RLE of the tung data

Figure 7.52: Cell-wise RLE of the tung data
qclust <- quickCluster(reads.qc, min.size = 30)
reads.qc <- computeSumFactors(reads.qc, sizes = 15, clusters = qclust)
reads.qc <- normalize(reads.qc)
plotPCA(
reads.qc[endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.53: PCA plot of the tung data after LSF normalisation

Figure 7.54: Cell-wise RLE of the tung data

Figure 7.55: Cell-wise RLE of the tung data
logcounts(reads.qc) <- log2(Down_Sample_Matrix(counts(reads.qc)) + 1)
plotPCA(
reads.qc[endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.56: PCA plot of the tung data after downsampling

Figure 7.57: Cell-wise RLE of the tung data

Figure 7.58: Cell-wise RLE of the tung data
reads.qc <- getBMFeatureAnnos(
reads.qc,
filters = "ensembl_gene_id",
attributes = c(
"ensembl_gene_id",
"hgnc_symbol",
"chromosome_name",
"start_position",
"end_position"
),
biomart = "ENSEMBL_MART_ENSEMBL",
dataset = "hsapiens_gene_ensembl",
host = "www.ensembl.org"
)tmp <- runPCA(
reads.qc.ann,
exprs_values = "tpm",
)
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
Figure 7.59: PCA plot of the tung data after TPM normalisation
7.10 处理混淆因子
7.10.1 介绍
在上一章中,我们对文库大小进行标准化,将其作为混淆因子移除。现在考虑从数据中删除其他混淆因子。 技术混淆因子(又称批次效应)可能源于试剂,分离方法,实验室/实验者,甚至实验的实施日期/时间的差异。 考虑技术混淆因子和批次效应是一个涉及实验设计原则的大课题。 在这里,我们讨论在实验设计合理时考虑混淆因子的方法。
从根本上说,解释技术混淆因子涉及识别并且理想地去除表达数据中与感兴趣的生物信号无关(即混淆)的变异来源。 目前的方法一些使用spike-in或管家基因,一些使用内源基因。
7.10.1.1 使用spike-ins去除混淆因子的优缺点
使用spike-ins作为对照基因是有吸引力的,因为在实验中向每个细胞添加相同量的ERCC(或其他)spike-ins。原则上,观察到的这些基因的所有变异都是由技术噪声引起的;而内源基因受技术噪声和生物变异性的影响。通过拟合spike-ins模型,可以从内源基因中去除技术噪声。基于此前提可以使用多种方法(例如 BASiCS, scLVM, RUVg);每个使用不同的噪声模型和不同的拟合程序。或者,可以识别出除技术噪声之外差异最显著的基因(例如,与中位数的距离, 高度可变基因)。然而,使用spike-ins进行标准化存在问题(特别是ERCC,源自细菌序列),包括变异实际上可以高于内源基因。
鉴于使用spike-ins的问题,使用内源基因获得更好的结果。在有大量内源基因的情况下,平均而言,细胞之间差异不明显,技术效应影响大量基因(这是一种非常普遍和合理的假设),那么这些方法(例如, RUVs方法)可以很好地应用于这种情况。
我们将探讨以下两种方法。
library(scRNA.seq.funcs)
library(RUVSeq)
library(scater)
library(SingleCellExperiment)
library(scran)
library(kBET)
library(sva) # Combat
library(edgeR)
library(harmony)
set.seed(1234567)
options(stringsAsFactors = FALSE)
umi <- readRDS("data/tung/umi.rds")
umi.qc <- umi[rowData(umi)$use, colData(umi)$use]
endog_genes <- !rowData(umi.qc)$is_feature_control
erccs <- rowData(umi.qc)$is_feature_control
qclust <- quickCluster(umi.qc, min.size = 30)
umi.qc <- computeSumFactors(umi.qc, sizes = 15, clusters = qclust)
umi.qc <- normalize(umi.qc)7.10.2 移除不需要的变异
导致技术噪声的因素经常表现为“批次效应”,其中在不同时间或不同技术人员处理的细胞彼此不同。通常可以使用相同或类似的工具来移除技术噪音并校正批次效应。我们使用 Remove Unwanted Variation (RUVSeq)。简而言之,RUVSeq的工作原理如下。对于\(n\)个样本和\(J\) 个基因,考虑以下广义线性模型(GLM),其中RNA-Seq reads counts在已知的感兴趣的协变量和不需要的变异的未知因子上回归: \[\log E[Y|W,X,O] = W\alpha + X\beta + O\] 这里,\(Y\)是观测到的基因水平read counts \(n \times J\) 矩阵,\(W\)是\(n \times k\)矩阵,对应“unwanted variation”因子,\(O\) 是\(n \times J\)偏移量矩阵,设置为0或者用其它标准化方法估计(比如上四分位数标准化)。同时估计\(W\), \(\alpha\), \(\beta\), 和 \(k\)是不可行的,给定k,使用以下三种方法估计“unwanted variation” \(W\):
- RUVg 使用阴性对照基因(比如,ERCCs),假设其在不同样本恒定表达;
- RUVs 使用中心化的(技术)重复/阴性对照样品,其中感兴趣的协变量是恒定的;
- RUVr 使用残差,比如来自感兴趣的协变量counts的first pass 广义线性回归。
我们关注前两种方法。
7.10.2.1 RUVg
7.10.2.2 RUVs
scIdx <- matrix(-1, ncol = max(table(umi.qc$individual)), nrow = 3)
tmp <- which(umi.qc$individual == "NA19098")
scIdx[1, 1:length(tmp)] <- tmp
tmp <- which(umi.qc$individual == "NA19101")
scIdx[2, 1:length(tmp)] <- tmp
tmp <- which(umi.qc$individual == "NA19239")
scIdx[3, 1:length(tmp)] <- tmp
cIdx <- rownames(umi.qc)
ruvs <- RUVs(counts(umi.qc), cIdx, k = 1, scIdx = scIdx, isLog = FALSE)
assay(umi.qc, "ruvs1") <- log2(
t(t(ruvs$normalizedCounts) / colSums(ruvs$normalizedCounts) * 1e6) + 1
)
ruvs <- RUVs(counts(umi.qc), cIdx, k = 10, scIdx = scIdx, isLog = FALSE)
assay(umi.qc, "ruvs10") <- log2(
t(t(ruvs$normalizedCounts) / colSums(ruvs$normalizedCounts) * 1e6) + 1
)7.10.3 Combat
如果你有一个平衡设计的实验,Combat可以用来消除批次效应的同时,通过使用mod参数指定生物效应来保留生物效应。 然而,Tung数据包含多个实验重复而不是平衡设计,因此使用mod1来保持生物变异将导致错误。
combat_data <- logcounts(umi.qc)
mod_data <- as.data.frame(t(combat_data))
# Basic batch removal
mod0 = model.matrix(~ 1, data = mod_data)
# Preserve biological variability
mod1 = model.matrix(~ umi.qc$individual, data = mod_data)
# adjust for total genes detected
mod2 = model.matrix(~ umi.qc$total_features_by_counts, data = mod_data)
assay(umi.qc, "combat") <- ComBat(
dat = t(mod_data),
batch = factor(umi.qc$batch),
mod = mod0,
par.prior = TRUE,
prior.plots = FALSE
)练习 1
将全部特征当做co-variate,进行ComBat校正,将校正后的矩阵存储在combat_tf slot中。
7.10.4 mnnCorrect
mnnCorrect (Haghverdi et al. 2017)假设每个批次和其它批次至少有一个生物学条件相同。因此,它适用于各种平衡的实验设计。 但是,Tung数据包含个体的多个重复而非平衡批次,因此我们分别对每个个体进行标准化。 请注意,由于实验设计混乱,这将去除同一个体内批次之间的批次效应,但不会去除不同个体批次之间的批次效应。
合并个体的重复以形成三个批次。
do_mnn <- function(data.qc) {
batch1 <- logcounts(data.qc[, data.qc$replicate == "r1"])
batch2 <- logcounts(data.qc[, data.qc$replicate == "r2"])
batch3 <- logcounts(data.qc[, data.qc$replicate == "r3"])
if (ncol(batch2) > 0) {
x = mnnCorrect(
batch1, batch2, batch3,
k = 20,
sigma = 0.1,
cos.norm.in = TRUE,
svd.dim = 2
)
res1 <- x$corrected[[1]]
res2 <- x$corrected[[2]]
res3 <- x$corrected[[3]]
dimnames(res1) <- dimnames(batch1)
dimnames(res2) <- dimnames(batch2)
dimnames(res3) <- dimnames(batch3)
return(cbind(res1, res2, res3))
} else {
x = mnnCorrect(
batch1, batch3,
k = 20,
sigma = 0.1,
cos.norm.in = TRUE,
svd.dim = 2
)
res1 <- x$corrected[[1]]
res3 <- x$corrected[[2]]
dimnames(res1) <- dimnames(batch1)
dimnames(res3) <- dimnames(batch3)
return(cbind(res1, res3))
}
}
indi1 <- do_mnn(umi.qc[, umi.qc$individual == "NA19098"])
indi2 <- do_mnn(umi.qc[, umi.qc$individual == "NA19101"])
indi3 <- do_mnn(umi.qc[, umi.qc$individual == "NA19239"])
assay(umi.qc, "mnn") <- cbind(indi1, indi2, indi3)
# For a balanced design:
#assay(umi.qc, "mnn") <- mnnCorrect(
# list(B1 = logcounts(batch1), B2 = logcounts(batch2), B3 = logcounts(batch3)),
# k = 20,
# sigma = 0.1,
# cos.norm = TRUE,
# svd.dim = 2
#)7.10.5 GLM
general linear model是“Combat”的简单版本。如果实验均衡设计,它可以校正批次效应同时保留生物效应。在混淆/重复实验设计中,生物效应将不适合/保留。 与mnnCorrect类似,可以分别从每个个体中去除批次效应,以保持个体之间的生物(和技术)差异。 下面仅出于演示目的,我们将纠正所有样本的批次效应:
For demonstation purposes we will naively correct all cofounded batch effects:
glm_fun <- function(g, batch, indi) {
model <- glm(g ~ batch + indi)
model$coef[1] <- 0 # replace intercept with 0 to preserve reference batch.
return(model$coef)
}
effects <- apply(
logcounts(umi.qc),
1,
glm_fun,
batch = umi.qc$batch,
indi = umi.qc$individual
)
corrected <- logcounts(umi.qc) - t(effects[as.numeric(factor(umi.qc$batch)), ])
assay(umi.qc, "glm") <- corrected练习2
对每个个体分别进行GLM校正,将校正后的矩阵存储于glm_indislot中。
glm_fun1 <- function(g, batch) {
model <- glm(g ~ batch)
model$coef[1] <- 0 # replace intercept with 0 to preserve reference batch.
return(model$coef)
}
do_glm <- function(data.qc) {
effects <- apply(
logcounts(data.qc),
1,
glm_fun1,
batch = data.qc$batch
)
corrected <- logcounts(data.qc) - t(effects[as.numeric(factor(data.qc$batch)), ])
return(corrected)
}
indi1 <- do_glm(umi.qc[, umi.qc$individual == "NA19098"])
indi2 <- do_glm(umi.qc[, umi.qc$individual == "NA19101"])
indi3 <- do_glm(umi.qc[, umi.qc$individual == "NA19239"])
assay(umi.qc, "glm_indi") <- cbind(indi1, indi2, indi3);7.10.6 Harmony
Harmony (Korsunsky and others 2018)是一个新的批次校正方法,在主成分空间中运行。该算法迭代聚类细胞,其目标函数定义成促进每个cluster包含不同数据集的细胞。 在聚类的基础上获得每个数据集的质心的位置,并校正坐标。 迭代此过程直到收敛。 Harmony带有一个控制批次校正程度的“theta”参数(值越高,导致更多的数据集整合),并且考虑输入的多个实验和生物因素。
查看Harmony的最终结果是如何在PCA的基础上创建降维空间,绘制所获得的流形并将其从该部分的后续分析中排除。
umi.qc.endog = umi.qc[endog_genes,]
umi.qc.endog = runPCA(umi.qc.endog, exprs_values = 'logcounts', ncomponents = 20)
pca <- as.matrix(umi.qc.endog@reducedDims@listData[["PCA"]])
harmony_emb <- HarmonyMatrix(pca, umi.qc.endog$batch, theta=2, do_pca=FALSE)
umi.qc.endog@reducedDims@listData[['harmony']] <- harmony_emb
plotReducedDim(
umi.qc.endog,
use_dimred = 'harmony',
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
7.10.7 评价和比较混淆因子去除策略
在考虑去除混淆因子的不同方法时,一个关键问题是如何定量地确定哪一种最有效。 比较具有挑战性的主要原因是因为通常很难知道那些对应技术混淆因子和什么是有趣的生物变异。根据对实验设计的知识,考虑三种不同的指标。 根据希望解决的生物学问题,选择在特定情况下评估影响最大的混淆因子指标。 选择一个允许您评估可能是特定情况下最大问题的混杂因素的指标非常重要。
7.10.7.1 效力标准1
通过检查PCA图来评估标准化的有效性,其中颜色对应于技术重复,形状对应于不同的生物样品(个体)。生物样品分离和散布的批次表明技术变异已被去除。 使用log2-cpm标准化数据。
for(n in assayNames(umi.qc)) {
tmp <- runPCA(
umi.qc[endog_genes, ],
exprs_values = n
)
print(
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
) +
ggtitle(n)
)
}











练习3
尝试RUV标准化不同的ks,哪个效果好?
7.10.7.2 效力标准2
还可以使用细胞间relative log expression(RLE)来确认技术噪声从数据集中删除来检查校正的效果。注意RLE仅评估每个细胞的高于和低于平均值的基因数量是否相等, 比如系统技术效应。RLE无法检测到不同批次间随机技术噪声。
res <- list()
for(n in assayNames(umi.qc)) {
res[[n]] <- suppressWarnings(calc_cell_RLE(assay(umi.qc, n), erccs))
}
par(mar=c(6,4,1,1))
boxplot(res, las=2) #### 效力标准3
#### 效力标准3
检查批次效应校正的另一种方法是考虑在数据的局部子样本中来自不同批次的点的混合。 如果没有批次效应,则任何局部区域中每批次的细胞比例应等于每批次中细胞的全局比例。
kBET (Buttner et al. 2017) 构建随机细胞的kNN网络,检验每个批次的细胞数目是否服从二项分布。 这些测试的拒绝率表明批次效应的严重性仍然存在于数据中(高拒绝率=强批次效应)。 kBET假设每个批次包含相同的complement of biological groups,因此如果使用了完美平衡的设计,它只能应用于整个数据集。但是,如果分别应用于每个生物组,“kBET”也可以应用于重复数据。 在Tung数据的情况下,我们分别将kBET应用于每个个体以检查残余批次效应。 然而,该方法不能识别与生物条件混淆的残留批次效应。 此外,kBET不能确定是否保留了生物信号。
compare_kBET_results <- function(sce){
indiv <- unique(sce$individual)
norms <- assayNames(sce) # Get all normalizations
results <- list()
for (i in indiv){
for (j in norms){
tmp <- kBET(
df = t(assay(sce[,sce$individual== i], j)),
batch = sce$batch[sce$individual==i],
heuristic = TRUE,
verbose = FALSE,
addTest = FALSE,
plot = FALSE)
results[[i]][[j]] <- tmp$summary$kBET.observed[1]
}
}
return(as.data.frame(results))
}
eff_debatching <- compare_kBET_results(umi.qc)require("reshape2")
require("RColorBrewer")
# Plot results
dod <- melt(as.matrix(eff_debatching), value.name = "kBET")
colnames(dod)[1:2] <- c("Normalisation", "Individual")
colorset <- c('gray', brewer.pal(n = 9, "RdYlBu"))
ggplot(dod, aes(Normalisation, Individual, fill=kBET)) +
geom_tile() +
scale_fill_gradient2(
na.value = "gray",
low = colorset[2],
mid=colorset[6],
high = colorset[10],
midpoint = 0.5, limit = c(0,1)) +
scale_x_discrete(expand = c(0, 0)) +
scale_y_discrete(expand = c(0, 0)) +
theme(
axis.text.x = element_text(
angle = 45,
vjust = 1,
size = 12,
hjust = 1
)
) +
ggtitle("Effect of batch regression methods per individual") 练习4
练习4
为什么原始counts几乎没有批次效应?
7.10.8 大作业
使用tung数据的read counts执行相同的分析。使用tung / reads.rds文件加载读取SCE对象。分析完成后,将结果与我们的结果进行比较(下一章)。 此外,尝试其他标准化组合并比较结果。
7.11 处理Reads中混淆因子
library(scRNA.seq.funcs)
library(RUVSeq)
library(scater)
library(SingleCellExperiment)
library(scran)
library(kBET)
library(sva) # Combat
library(harmony)
library(edgeR)
set.seed(1234567)
options(stringsAsFactors = FALSE)
reads <- readRDS("data/tung/reads.rds")
reads.qc <- reads[rowData(reads)$use, colData(reads)$use]
endog_genes <- !rowData(reads.qc)$is_feature_control
erccs <- rowData(reads.qc)$is_feature_control
qclust <- quickCluster(reads.qc, min.size = 30)
reads.qc <- computeSumFactors(reads.qc, sizes = 15, clusters = qclust)
reads.qc <- normalize(reads.qc)ruvg <- RUVg(counts(reads.qc), erccs, k = 1)
assay(reads.qc, "ruvg1") <- log2(
t(t(ruvg$normalizedCounts) / colSums(ruvg$normalizedCounts) * 1e6) + 1
)
ruvg <- RUVg(counts(reads.qc), erccs, k = 10)
assay(reads.qc, "ruvg10") <- log2(
t(t(ruvg$normalizedCounts) / colSums(ruvg$normalizedCounts) * 1e6) + 1
)scIdx <- matrix(-1, ncol = max(table(reads.qc$individual)), nrow = 3)
tmp <- which(reads.qc$individual == "NA19098")
scIdx[1, 1:length(tmp)] <- tmp
tmp <- which(reads.qc$individual == "NA19101")
scIdx[2, 1:length(tmp)] <- tmp
tmp <- which(reads.qc$individual == "NA19239")
scIdx[3, 1:length(tmp)] <- tmp
cIdx <- rownames(reads.qc)
ruvs <- RUVs(counts(reads.qc), cIdx, k = 1, scIdx = scIdx, isLog = FALSE)
assay(reads.qc, "ruvs1") <- log2(
t(t(ruvs$normalizedCounts) / colSums(ruvs$normalizedCounts) * 1e6) + 1
)
ruvs <- RUVs(counts(reads.qc), cIdx, k = 10, scIdx = scIdx, isLog = FALSE)
assay(reads.qc, "ruvs10") <- log2(
t(t(ruvs$normalizedCounts) / colSums(ruvs$normalizedCounts) * 1e6) + 1
)combat_data <- logcounts(reads.qc)
mod_data <- as.data.frame(t(combat_data))
# Basic batch removal
mod0 = model.matrix(~ 1, data = mod_data)
# Preserve biological variability
mod1 = model.matrix(~ reads.qc$individual, data = mod_data)
# adjust for total genes detected
mod2 = model.matrix(~ reads.qc$total_features_by_counts, data = mod_data)
assay(reads.qc, "combat") <- ComBat(
dat = t(mod_data),
batch = factor(reads.qc$batch),
mod = mod0,
par.prior = TRUE,
prior.plots = FALSE
)练习1
assay(reads.qc, "combat_tf") <- ComBat(
dat = t(mod_data),
batch = factor(reads.qc$batch),
mod = mod2,
par.prior = TRUE,
prior.plots = FALSE
)do_mnn <- function(data.qc) {
batch1 <- logcounts(data.qc[, data.qc$replicate == "r1"])
batch2 <- logcounts(data.qc[, data.qc$replicate == "r2"])
batch3 <- logcounts(data.qc[, data.qc$replicate == "r3"])
if (ncol(batch2) > 0) {
x = mnnCorrect(
batch1, batch2, batch3,
k = 20,
sigma = 0.1,
cos.norm.in = TRUE,
svd.dim = 2
)
res1 <- x$corrected[[1]]
res2 <- x$corrected[[2]]
res3 <- x$corrected[[3]]
dimnames(res1) <- dimnames(batch1)
dimnames(res2) <- dimnames(batch2)
dimnames(res3) <- dimnames(batch3)
return(cbind(res1, res2, res3))
} else {
x = mnnCorrect(
batch1, batch3,
k = 20,
sigma = 0.1,
cos.norm.in = TRUE,
svd.dim = 2
)
res1 <- x$corrected[[1]]
res3 <- x$corrected[[2]]
dimnames(res1) <- dimnames(batch1)
dimnames(res3) <- dimnames(batch3)
return(cbind(res1, res3))
}
}
indi1 <- do_mnn(reads.qc[, reads.qc$individual == "NA19098"])
indi2 <- do_mnn(reads.qc[, reads.qc$individual == "NA19101"])
indi3 <- do_mnn(reads.qc[, reads.qc$individual == "NA19239"])
assay(reads.qc, "mnn") <- cbind(indi1, indi2, indi3)
# For a balanced design:
#assay(reads.qc, "mnn") <- mnnCorrect(
# list(B1 = logcounts(batch1), B2 = logcounts(batch2), B3 = logcounts(batch3)),
# k = 20,
# sigma = 0.1,
# cos.norm = TRUE,
# svd.dim = 2
#)glm_fun <- function(g, batch, indi) {
model <- glm(g ~ batch + indi)
model$coef[1] <- 0 # replace intercept with 0 to preserve reference batch.
return(model$coef)
}
effects <- apply(
logcounts(reads.qc),
1,
glm_fun,
batch = reads.qc$batch,
indi = reads.qc$individual
)
corrected <- logcounts(reads.qc) - t(effects[as.numeric(factor(reads.qc$batch)), ])
assay(reads.qc, "glm") <- corrected练习2
glm_fun1 <- function(g, batch) {
model <- glm(g ~ batch)
model$coef[1] <- 0 # replace intercept with 0 to preserve reference batch.
return(model$coef)
}
do_glm <- function(data.qc) {
effects <- apply(
logcounts(data.qc),
1,
glm_fun1,
batch = data.qc$batch
)
corrected <- logcounts(data.qc) - t(effects[as.numeric(factor(data.qc$batch)), ])
return(corrected)
}
indi1 <- do_glm(reads.qc[, reads.qc$individual == "NA19098"])
indi2 <- do_glm(reads.qc[, reads.qc$individual == "NA19101"])
indi3 <- do_glm(reads.qc[, reads.qc$individual == "NA19239"])
assay(reads.qc, "glm_indi") <- cbind(indi1, indi2, indi3);reads.qc.endog = reads.qc[endog_genes,]
reads.qc.endog = runPCA(reads.qc.endog, exprs_values = 'logcounts', ncomponents = 20)
pca <- as.matrix(reads.qc.endog@reducedDims@listData[["PCA"]])
harmony_emb <- HarmonyMatrix(pca, reads.qc.endog$batch, theta=2, do_pca=FALSE)
reads.qc.endog@reducedDims@listData[['harmony']] <- harmony_emb
plotReducedDim(
reads.qc.endog,
use_dimred = 'harmony',
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
for(n in assayNames(reads.qc)) {
tmp <- runPCA(
reads.qc[endog_genes, ],
exprs_values = n
)
print(
plotPCA(
tmp,
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
) +
ggtitle(n)
)
}











res <- list()
for(n in assayNames(reads.qc)) {
res[[n]] <- suppressWarnings(calc_cell_RLE(assay(reads.qc, n), erccs))
}
par(mar=c(6,4,1,1))
boxplot(res, las=2)
compare_kBET_results <- function(sce){
indiv <- unique(sce$individual)
norms <- assayNames(sce) # Get all normalizations
results <- list()
for (i in indiv){
for (j in norms){
tmp <- kBET(
df = t(assay(sce[,sce$individual== i], j)),
batch = sce$batch[sce$individual==i],
heuristic = TRUE,
verbose = FALSE,
addTest = FALSE,
plot = FALSE)
results[[i]][[j]] <- tmp$summary$kBET.observed[1]
}
}
return(as.data.frame(results))
}
eff_debatching <- compare_kBET_results(reads.qc)require("reshape2")
require("RColorBrewer")
# Plot results
dod <- melt(as.matrix(eff_debatching), value.name = "kBET")
colnames(dod)[1:2] <- c("Normalisation", "Individual")
colorset <- c('gray', brewer.pal(n = 9, "RdYlBu"))
ggplot(dod, aes(Normalisation, Individual, fill=kBET)) +
geom_tile() +
scale_fill_gradient2(
na.value = "gray",
low = colorset[2],
mid=colorset[6],
high = colorset[10],
midpoint = 0.5, limit = c(0,1)) +
scale_x_discrete(expand = c(0, 0)) +
scale_y_discrete(expand = c(0, 0)) +
theme(
axis.text.x = element_text(
angle = 45,
vjust = 1,
size = 12,
hjust = 1
)
) +
ggtitle("Effect of batch regression methods per individual")
## R version 3.6.0 (2019-04-26)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 7 x64 (build 7601) Service Pack 1
##
## Matrix products: default
##
## locale:
## [1] LC_COLLATE=Chinese (Simplified)_People's Republic of China.936
## [2] LC_CTYPE=Chinese (Simplified)_People's Republic of China.936
## [3] LC_MONETARY=Chinese (Simplified)_People's Republic of China.936
## [4] LC_NUMERIC=C
## [5] LC_TIME=Chinese (Simplified)_People's Republic of China.936
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] scRNA.seq.funcs_0.1.0 limma_3.41.6
## [3] scater_1.13.9 ggplot2_3.2.0
## [5] SingleCellExperiment_1.7.0 SummarizedExperiment_1.15.5
## [7] DelayedArray_0.11.4 BiocParallel_1.19.0
## [9] matrixStats_0.54.0 Biobase_2.45.0
## [11] GenomicRanges_1.37.14 GenomeInfoDb_1.21.1
## [13] IRanges_2.19.10 S4Vectors_0.23.17
## [15] BiocGenerics_0.31.5 knitr_1.23
##
## loaded via a namespace (and not attached):
## [1] viridis_0.5.1 BiocSingular_1.1.5
## [3] viridisLite_0.3.0 DelayedMatrixStats_1.7.1
## [5] elliptic_1.4-0 moments_0.14
## [7] assertthat_0.2.1 statmod_1.4.32
## [9] highr_0.8 GenomeInfoDbData_1.2.1
## [11] vipor_0.4.5 yaml_2.2.0
## [13] robustbase_0.93-5 pillar_1.4.2
## [15] lattice_0.20-38 glue_1.3.1
## [17] digest_0.6.20 XVector_0.25.0
## [19] colorspace_1.4-1 cowplot_1.0.0
## [21] htmltools_0.3.6 Matrix_1.2-17
## [23] pkgconfig_2.0.2 bookdown_0.12
## [25] zlibbioc_1.31.0 purrr_0.3.2
## [27] scales_1.0.0 Rtsne_0.15
## [29] tibble_2.1.3 withr_2.1.2
## [31] lazyeval_0.2.2 magrittr_1.5
## [33] crayon_1.3.4 evaluate_0.14
## [35] MASS_7.3-51.4 beeswarm_0.2.3
## [37] tools_3.6.0 stringr_1.4.0
## [39] munsell_0.5.0 irlba_2.3.3
## [41] orthopolynom_1.0-5 compiler_3.6.0
## [43] rsvd_1.0.1 contfrac_1.1-12
## [45] rlang_0.4.0 grid_3.6.0
## [47] RCurl_1.95-4.12 BiocNeighbors_1.3.2
## [49] bitops_1.0-6 labeling_0.3
## [51] rmarkdown_1.14 hypergeo_1.2-13
## [53] gtable_0.3.0 deSolve_1.23
## [55] R6_2.4.0 gridExtra_2.3
## [57] dplyr_0.8.3 stringi_1.4.3
## [59] ggbeeswarm_0.6.0 Rcpp_1.0.1
## [61] DEoptimR_1.0-8 tidyselect_0.2.5
## [63] xfun_0.8References
Anders, Simon, and Wolfgang Huber. 2010. “Differential Expression Analysis for Sequence Count Data.” Genome Biol 11 (10): R106. https://doi.org/10.1186/gb-2010-11-10-r106.
Bullard, James H, Elizabeth Purdom, Kasper D Hansen, and Sandrine Dudoit. 2010. “Evaluation of Statistical Methods for Normalization and Differential Expression in mRNA-Seq Experiments.” BMC Bioinformatics 11 (1): 94. https://doi.org/10.1186/1471-2105-11-94.
Buttner, Maren, Zhichao Miao, Alexander Wolf, Sarah A Teichmann, and Fabian J Theis. 2017. “Assessment of Batch-Correction Methods for scRNA-seq Data with a New Test Metric.” bioRxiv, October, 200345.
Haghverdi, Laleh, Aaron T L Lun, Michael D Morgan, and John C Marioni. 2017. “Correcting Batch Effects in Single-Cell RNA Sequencing Data by Matching Mutual Nearest Neighbours.” bioRxiv, July, 165118.
Korsunsky, I, and others. 2018. “Fast, Sensitive, and Accurate Integration of Single Cell Data with Harmony. BioRxiv.”
L. Lun, Aaron T., Karsten Bach, and John C. Marioni. 2016. “Pooling Across Cells to Normalize Single-Cell RNA Sequencing Data with Many Zero Counts.” Genome Biol 17 (1). https://doi.org/10.1186/s13059-016-0947-7.
Robinson, Mark D, and Alicia Oshlack. 2010. “A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data.” Genome Biol 11 (3): R25. https://doi.org/10.1186/gb-2010-11-3-r25.
Tung, Po-Yuan, John D. Blischak, Chiaowen Joyce Hsiao, David A. Knowles, Jonathan E. Burnett, Jonathan K. Pritchard, and Yoav Gilad. 2017. “Batch Effects and the Effective Design of Single-Cell Gene Expression Studies.” Sci. Rep. 7 (January): 39921. https://doi.org/10.1038/srep39921.